Возможные осложнения и прогноз
Исход недуга зависит от степени выраженности патологических изменений. Как правило, костные деформации не несут угрозы жизни и здоровью ребенка. Малыши не отстают в умственном развитии, однако испытывают трудности с социальной адаптацией, как из-за специфической внешности, так и в результате нарушений слуха и речи.
Опасными могут быть сочетающиеся с синдромом Фраческетти пороки сердца, а также дефекты костей, приводящие к затруднению дыхания пациента. В таких случаях прогноз более осторожный. Большинство проблем, возникающих на фоне этой генетической мутации, поддаются хирургическому лечению.
Специфических методов предупреждения развития синдрома Тричера Коллинза не разработано. Эта особенность связана с тем, что контролировать хромосомные мутации не удается. Поэтому профилактика возникновения дефектов направлена на правильное планирование беременности. Оно подразумевает проведение кариотипирования родителей с целью выявления хромосомных аномалий. Однако даже такой подход не дает гарантий отсутствия заболевания у будущего ребенка. Это связано с широкой распространенностью спонтанных мутаций, приводящих к формированию дизостоза.
* Факторы синдрома Трейчера Коллинза, под редакцией: Чарльз Патрик Дэвис, MD, PhD
- Синдром Трехара Коллинза (TCS) — это состояние (генетическое заболевание), которое изменяет развитие костей и других тканей в лице.
- Признаки и симптомы варьируются от почти незаметных изменений лица до тяжелых изменений лица и уха, расщелины неба и ограниченных дыхательных путей
- Характеристики TCS включают черепно-лицевые или мандибулофациальные аномалии:
- Глаза, которые уклоняются от носа
- Очень немного ресниц и выемки в нижних веках (глаз coloboma)
- Уши, которые отсутствуют или необычно сформированы
- У некоторых людей может быть потеря слуха
- Маленькая челюсть
- Ребенок с ТКС может иметь апноэ во сне и / или проводящую потерю слуха; для потери слуховой функции может потребоваться ресурс для предоставления слуховых аппаратов для детей.
- Некоторые люди могут серьезно пострадать, и у них могут развиться опасные для жизни проблемы с дыханием (инфантильное апноэ).
- Другие аномалии могут затруднить дыхание и кормление ребенка из-за суженной обструкции носовых дыхательных путей.
- У ребенка могут быть особенности «последовательности Пьера Робена», в которой язык находится дальше в горле, чем обычно (глоссоптоз), с или без, и неполное расщепление неба рта и обструкции дыхательных путей.
Харри Реймонд Истлек
list25.com
Харри страдал от очень редкого недуга под названием прогрессирующая оссифицирующая фибродисплазия (ФОП). При этом у человека соединительные ткани начинают превращаться в кости, в результате чего формируется буквально новая структура скелета.
Уровни развития недуга
В начале статьи было указано, что существует несколько стадий болезни, о которых пришло время поговорить подробнее. Синдром Франческетти (Коллинза) имеет три уровня развития. Первый и наиболее безопасный характеризуется небольшой гипоплазией костей лицевой части. Однако даже в этом случае человек меняется внешне, и самые внимательные могут заподозрить наличие заболевания.
Когда наступает вторая стадия, то здесь уже добавляются проблемы со слухом, неправильная форма глазных щелей, деформация нижней челюсти. В этом случае недуг становится более явным, и человек с синдромом внешне значительно отличается от обычного индивида.
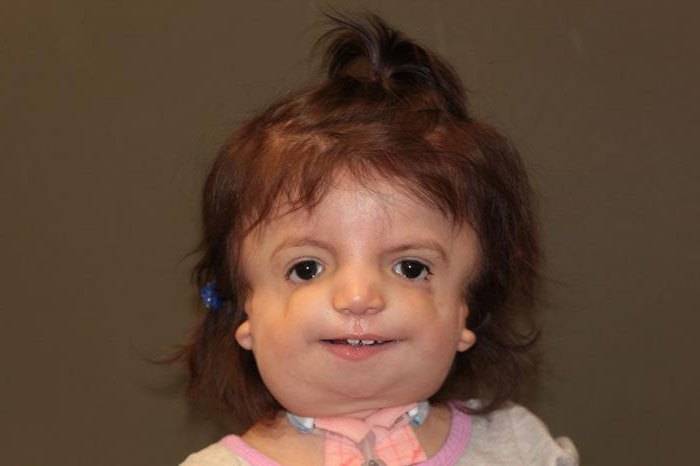
Что касается последней, наиболее тяжёлой стадии, то тут имеет место полное отсутствие лица. На самом деле это очень страшно, ведь пациент просто автоматически исключается из общества, и с очень низкой долей вероятности что-то изменится. Кроме того, эти люди с годами страдают сильнее, ведь патология только усугубляется. Могут появиться признаки более тяжёлых болезней. Синдром Франческетти – сам по себе довольно опасный недуг, а с годами могут приобретаться заболевания и похуже.
Последствия синдрома Тричера Коллинза
Генетическая патология костей черепа и лица влечёт массу опасных последствий, таких как:
- Тугоухость (вызванная недоразвитостью слуховых костей или отсутствием ушных раковин).
- Косоглазие.
- Различного рода стоматологические заболевания.
- Невозможность самостоятельного приёма пищи (вследствие деформации костей челюсти и редком и/или неправильном расположении зубов, отсутствии слюнных желёз).
- Нарушения дыхательной функции (из-за зарастания носовых ходов, аномального развития верхнего нёба и увеличенного в размерах, «выпихиваемого» языка, который может заблокировать дыхательные пути).
В редких случаях синдром становится причиной заболеваний сердечно-сосудистой системы и мутации некоторых других внутренних органов.
Информация
Синдром Тричера Коллинза – редкое генетическое заболевание, встречающееся у лиц обоих полов. В особо тяжёлых случаях, оно характеризуется практически полным отсутствием лица и лицевых костей. Вылечить патологию невозможно, но большинство мутаций можно значительно уменьшить за счёт сложных, многоступенчатых хирургических операций. Лечение проводится комплексно, включая занятия с логопедом, психологом.
Зачастую уродства лица у детей обусловлены генетической патологией. Среди таких – синдром Тричера-Коллинза. Заболевание сопровождается деформацией костей черепа, лицевого отдела. При отсутствии медицинской помощи болезнь быстро прогрессирует.
Синдром Тричера-Коллинза – что это такое?
Рассказывая про синдром Тричера-Коллинза, что это за патология, медики нередко используют другое название – синдром Франческетти. Данной патологией принято обозначать комплекс нарушений, затрагивающих деформацию костей черепа. Заболевание протекает длительно, поэтому в течении болезни принято выделять стадии. По частоте встречаемости синдром относят к редким генетическим патологиям: заболевание регистрируется с частотой 1 случай на 10 000 младенцев и является наследственным.
Синдром Тричера-Коллинза – причины
В начале XX века впервые был описан синдром Тричера-Коллинза: причины заболевания тогда достоверно установить не удалось. В ходе длительных исследований ученые выяснили, что патология развивается в результате изменений структуры ДНК. Мутации развиваются в 5 хромосоме. Она является одной из самых длинных нуклеотидных структур в составе человеческого генома и ответственна за правильное формирование скелета у зародыша.
Провоцирующим фактором, вызывающим синдром Тричера-Коллинза (фото изображено ниже), является сбой внутриклеточного синтеза белка. В итоге развивается синдром гаплонедостаточности – нехватки белка, необходимого для нормального развития лицевой части черепа. Стоит отметить, что патология может носить как наследственный, так и приобретенный характер.
Челюстно-лицевой дизостоз – тип наследования
Генетики, описывая челюстно-лицевой дизостоз, тип наследования болезни, указывают аутосомно-доминантный. Это означает, что патология развивается, если у одного из родителей есть мутантный ген. В таком случае мутации затрагивают гены TCOF1 или POLR1D. Однако по наблюдениям специалистов, синдром Тричера-Коллинза развивается и при мутации собственных генов в организме пациента.
Наличие мутации гена POLR1C подтверждает этот факт. В редких случаях заболевание Тричера-Коллинза передается по аутосомно-рецессивному типу наследования, когда ребенок получает мутировавший ген от обоих родителей. При этом патология у мамы или папы может практически не проявляться или иметь едва заметные симптомы.

Возможные осложнения и прогноз
Если заболевание не сопровождается грубыми пороками развития внутренних органов, то прогноз для жизни и здоровья ребенка в будущем благоприятен. При наличии тугоухости снижается способность к развитию речи, письма, обучению.
Необычная внешность также мешает получению навыков социального общения, так как другие дети стараются избегать сверстников с врожденными аномалиями и уродствами. В результате у ребенка может быть значительно снижена самооценка, что требует консультации со стороны психолога. Проведение ряда пластических операций позволяет частично сгладить дефекты развития.
Тугоухость часто ошибочно диагностируется как недоразвитие психики. Умственная отсталость у таких детей встречается редко, а некоторые имеют способности выше среднего
Поэтому у таких пациентов важно на ранней стадии выявлять нарушения слуха и корректировать их
Врачи рекомендуют это делать в обязательном порядке до достижения ребенком шестимесячного возраста. С 3-х месяцев он может носить аппарат для костного проведения звуковых волн, а после 3 лет возможна установка имплантата за ушами. Также очень большое значение имеют занятия с сурдопедагогом.
В первые месяцы жизни недоразвитость нижней челюсти может способствовать выпадению языка и перекрытию дыхательных путей, что является потенциально опасным для жизни ребенка. В последующем могут возникнуть затруднения в приеме пищи из-за ограниченной возможности открытия рта (в различной степени тяжести).
Лечение
В случае, когда заболевание диагностировано еще во время внутриутробного развития, женщине рекомендуют сделать аборт. В противном случае, сразу после рождения ребенку потребуется квалифицированное комплексное лечение по устранению дефектов, насколько это возможно. Терапия заключается в следующих действиях:
- Хирургическое вмешательство, в ходе которого устраняются дефекты лица. Обычно необходимо несколько операций, чтобы уменьшить проявления болезни. Все зависит от сложности случая.
- Стоматологические процедуры. Обычно у ребенка диагностируют не только проблемы с зубами, но и не правильный прикус, которые с помощью дантистов можно корректировать.
- Чтобы улучшить костнопроводимость звуков, потребуется установить слуховые аппараты.
Если у ребенка наблюдаются проблемы с дыханием и питанием, то потребуются операции по решению этих проблем (трахеотомия и гастростомия). Для дальнейшей адаптации ребенка потребуется работа с логопедами и сурдопедагогами.
Профилактика и прогноз
СТК — пожизненный диагноз. Заболевание протекает достаточно тяжело и требует высококвалифицированной помощи. Поскольку его причиной является генетическая мутация, предупредить развитие синдрома невозможно. Медико-генетическое консультирование требуется парам с негативным семейным анамнезом. Если данное заболевание не было зафиксировано у близких и дальних родственников, необходимо соблюдать стандартные рекомендации относительно здорового образа жизни во время беременности.
Причины синдрома Апера
К основным причинам мутации генов, вызывающих впоследствии нарушения в формировании костной ткани и костных соединений, можно отнести:
- перенесенные заболевания матери при беременности (сифилис, грипп, туберкулез, краснуха, менингит);
- рентгеновское облучение женщины в период беременности, особенно опасна данная процедура на ранних стадиях;
- рождение ребенка в пожилом возрасте повышает шансы проявления у младенца генетических мутаций;
- генетическая предрасположенность, наличие в роду подобных генетических патологий.
Наследование заболевания составляет около 50% и абсолютно не зависит от пола новорожденного. По статистике, генетическая мутация встречается у одного из двадцати тысяч детей.
На сковородке с хрустящей корочкой
Стадии
Стадии синдрома определяются сложностью мутационного процесса и интенсивностью клинических признаков:
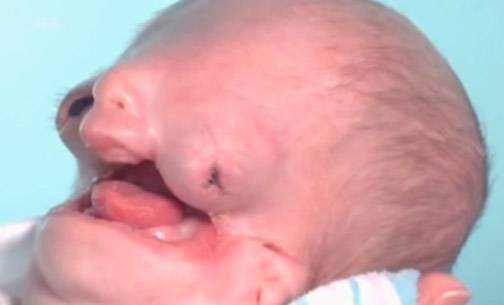
СТК тяжелой степени
- Начальная стадия характеризуется практически незаметными изменениями на лице. Больные дети ни чем не отличаются от здоровых и ведут нормальный образ жизни.
- Средняя стадия проявляется всеми перечисленными выше нарушениями. Аномальная деформация лицевых костей достаточно сильная. Возможны трудности с дыханием, принятием пищи, нарушение слуха, проблемы с зубами.
- Тяжелая стадия — полное отсутствие лица, невозможность рассмотреть его черты. Даже пластическая хирургия не может помочь больным.
Стадии
Учитывая всю проблематичность мутаций, проявления данного недуга могут быть выражены очень ярко, или же быть незначительными.
Классифицируют такие стадии патологического процесса:
- На первом этапе видоизменения практически незаметны. Такие пациенты счастливчики, ведь они попали именно в ту категорию, которая позволяет им вести полноценный образ жизни.
- На второй стадии признаки налицо, как в прямом, так и в переносном смысле. Также могут возникнуть и сложности с функцией дыхания и функционированием других органов.
- Одной из наиболее сложных является третья стадия. Она сопровождается почти полной деформацией лица. При этом никакая терапия и пластика не будут эффективны.
Синдром Тричера Коллинза: причины болезни и лечение заболевания
Синдромом Тричера-Коллинза называют заболевание наследственного характера, характеризующееся деформацией костных структур черепа и лица. Другое название болезни – челюстно-лицевой дизостоз.
Патология возникает у одного новорожденного на 50 тысяч малышей. Подробнее о синдроме Тричера-Коллинза и жизни людей, имеющих дефекты на уровне генов, рассказано в статье.
Синдром Тричера-Коллинза: причины заболевания
Эдвард Тричер Коллинз описал основные нарушения, возникающие у больных людей, более ста лет назад. Патология получила соответствующее название. Современная медицина предпочитает использовать другие термины – мандибулофасциальный дизостоз или синдром Франческетти-Коллинза.
Основным фактором, который приводит к развитию заболевания, считается врожденная аномалия в гене 5-ой хромосомы.
Этот ген считается ответственным за кодировку белка, принимающего участие в правильном формировании костей лицевой части черепа в период внутриутробного развития ребенка. Белок называется ядрышковым фосфопротеином.
В конце первого месяца беременности лицевой отдел плода заполняется мезенхимальными тканями, которые на протяжении второго месяца уже дифференцируются в кости и соединительнотканные элементы.
Заболевание может возникнуть в случае тератогенного влияния следующих факторов:
- этанол и его производные;
- радиоактивное излучение;
- цитомегаловирус;
- токсоплазмоз;
- влияние гербицидов;
- препараты на основе ретиноевой кислоты;
- противосудорожные лекарства;
- психотропные препараты.
Симптомы патологии
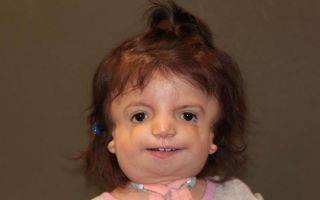
Основными проявлениями синдрома Тричера-Коллинза считаются:
- опущенные углы глаз;
- недоразвитость скуловых костей с обеих сторон;
- отсутствие части тканей века;
- недоразвитость кости челюсти;
- отсутствие или малый размер раковин ушей;
- отсутствие или заращение наружных слуховых проходов врожденного характера;
- анатомические дефекты среднего отдела уха;
- «птичье лицо».
Так как височная кость является сложным элементом черепно-лицевого отдела, в некоторых случаях всю тяжесть поражений анатомо-физиологических особенностей наружного слухового прохода и внутренних структур уха оценить достаточно трудно.
Визуальный осмотр больного человека позволяет определить отсутствие ресниц на нижнем веке, в некоторых случаях – расщелину неба. При синдроме Тричера-Коллинза возможны врожденные дефекты и нарушения работы со стороны сердца и элементов опорно-двигательного аппарата (помимо лицевого отдела).
Особенности жизни человека с синдромом Тричера-Коллинза можно увидеть в документальном фильме «Новое лицо Джулианы».
На момент одиннадцати лет ребенок перенес уже более 50 операций, но лечение еще продолжается.
Родители Джулианы имеют также старшую здоровую дочь. Они хотели еще одного ребенка, однако, побоялись его планировать из-за возможности передачи генной мутации. Взамен родители удочерили украинскую девочку, имеющую такую же генетическую болезнь, но в менее тяжелом проявлении.
Лечение синдрома Тричера-Коллинза
Это заболевание, как и множество других генетических дефектов и нарушений, лечению не поддается. Используется паллиативная терапия, которая позволяет поддержать жизнь больного на достаточном уровне.
Для коррекции слуховых функций используют слуховые аппараты, а чтобы улучшить речь, необходима помощь специалиста-логопеда.
Показан ряд и других пластических операций для лечения и восстановления анатомических дефектов.
Джон Ланкастер и синдром Тричера-Коллинза
Джон Ланкастер – парень, проживающий в Великобритании. На данный момент ему 26 лет. Биологические родители отказались от Джона сразу после его рождения, они не смогли смириться с грозным заболеванием, однако ребенка усыновила другая пара, которая смогла обеспечить его всем необходимым.
Ланкастер перенес несколько пластических операций, которые немного изменили его внешность.
На данный момент он показывает своим примером, что даже с генетическими аномалиями можно жить и радоваться жизни.
Сестра Аделины Сотниковой и синдром Тричера-Коллинза
Аделина Сотникова – российская олимпийская чемпионка по фигурному катанию. Ее младшая сестра Маша имеет генетическое заболевание, о чем стало известно недавно, после победы Аделины в Сочи.
После рождения младшей сестры родителей Аделины стали готовить в роддоме к тому, что больного синдромом Тричера-Коллинза ребенка придется оставить, но они отказались, приняв решение заботиться о дочери и бороться за ее здоровье.
Свои гонорары Аделина Сотникова тратила на лечение сестры Маши. Девушке еще предстоят хирургические вмешательства, поскольку она продолжает расти.
Этиология и патогенез
Единственным этиопатогенетическим фактором синдрома Тричера Коллинза считается генетическая мутация. Врожденная аномалия структуры пятой хромосомы является причиной недуга. Это самая длинная нуклеотидная структура в геноме человека, отвечающая за производство материала для скелета плода. В организме больного нарушается биогенез и функции рибосомной РНК, происходит сбой внутриклеточного синтеза белка, замедляется процесс деления эмбриональных клеток нервной трубки. Их самоуничтожение приводит к недоразвитию костной ткани и формированию обезображенного лица ребенка на раннем этапе эмбриогенеза. Диагностировать недуг можно уже на втором месяце беременности.
Синдром в 100% случаев наследуется от больной матери или отца по доминантному принципу. Если имеется отягощенный семейный анамнез, то в данной семье с этой патологией обязательно рождаются больные дети. Экспрессивность и пенетрантность гена обуславливают различную степень выраженности дефекта, которая варьируется у разных пациентов от умеренной до крайне тяжелой.
В некоторых случаях синдром не наследуется, а формируется в силу новой мутации генов после зачатия. Рождаются дети с синдромом от абсолютно здоровых родителей. Мутация может произойти под влиянием факторов, оказывающих тератогенное воздействие на плод:
- злоупотребление беременной женщиной алкоголем,
- курение и наркотическая зависимость,
- сильный стресс,
- вирусные и бактериальные инфекции у женщины,
- тяжелые сопутствующие патологические процессы,
- применение определенных лекарств — психотропных и противосудорожных препаратов,
- радиационное облучение.
Лечебные мероприятия
Как и для большинства генетических заболеваний, специального лечения при данном синдроме не существует. Терапия представляет собой сложную, многопрофильную задачу и зависит от тяжести поражения и осложнений.
Проводятся следующие лечебные мероприятия:
- В случае дыхательной недостаточности – трахеостомия. Эта процедура заключается в создании проходимости в трахее с помощью установки трубки для обеспечения поступления воздуха в дыхательные пути. В легких случаях возможна неинвазивная искусственная вентиляция легких.
- Дистракция (растяжение) нижней челюсти хирургическим путем с помощью специального аппарата. Необходимость проведения такой манипуляции рассматривается врачами в индивидуальном порядке.
- Установка гастростомы – трубки, которая требуется для защиты дыхательных путей от попадания пищи.
- Хирургическая реконструкция лицевой части черепа. Она обычно проводится с 5-6 лет.
При наличии осложнений показаны консультации у соответствующих специалистов и патогенетическое лечение.
Хирургические способы восстановления слуха у таких детей неэффективны. Рекомендуемая тактика реабилитации – использование слуховых аппаратов костной проводимости (или обычных слуховых аппаратов при незначительной деформации ушной раковины).
Их особенностью является то, что звуки передаются во внутреннее ухо по костям черепа. Это не так физиологично, как воздушное звукопроведение, однако при определенном усилении они очень хорошо воспринимаются рецепторами.
При данном заболевании хорошо зарекомендовали себя имплантируемые слуховые аппараты ВАНА (БАХА), которые имеют в своем составе титановую опору, закрепляемую в толще височной кости. Со временем титан срастается с костной тканью, а штифт напрямую передает звуковые колебания в улитку внутреннего уха.
Хирургическая установка имплантата производится в 2 этапа: сначала внедряется титановый штифт, а затем, после его вживления в кость в течение полугода, монтируется опора. Через месяц на нее надевают звуковой процессор.
Такие слуховые аппараты обладают следующими преимуществами:
- у детей отмечается улучшение порогов слышимости в диапазоне громкости обычной речи;
- по сравнению с обычными аппаратами и хирургической реконструкцией эстетические и аудиологические показатели выше;
- происходит спонтанное улучшение речи и постановки голоса (его высоты и интенсивности).
У маленьких детей слухопротезирование проводится с применением эластичной ленты, закрепляемой на голове. Существуют также цифровые аппараты костного проведения звуков, имплантируемые в височную кость (Alpha).
Возможные осложнения и прогноз
Вероятность развития осложнений на фоне отсутствия хирургического лечения высока. К наиболее вероятным из них относятся:
- развитие слабоумия;
- снижение зрения и слуха;
- заболевания позвоночника и суставов;
- патологии сердечно-сосудистой, выделительной и других систем.
Прогноз при отсутствии операции неблагоприятный, за исключением тех пациентов, которые дожили до средних лет, не имея пороков развития сердечно-сосудистой системы.
В случае своевременно проведённых оперативных вмешательств и лёгкой степени развития патологии пациент может жить до старости при условии, что мутация гена не повлекла за собой пороки сердца и тяжёлые патологии внутренних органов.
Диагностика во время беременности
Диагностика заболевания плода в период его вынашивания производится 2 способами: с помощью ультразвукового исследования, на котором выявляются грубые челюстно-лицевые нарушения, и молекулярного исследования образцов биологического материала.
Молекулярно-генетическая диагностика позволяет выявить изменения в ответственном гене путем прямого определения аминокислотной последовательности, которое производится автоматически.
В качестве биологического материала используются:
- образцы ворсин внешней оболочки зародыша, если срок беременности находится в пределах 8-14 недель;
- околоплодные воды, если женщина беременна на сроке 16-21 недель.
Стоимость исследования составляет порядка 30 тыс. руб., срок выполнения около 1 месяца, цена поиска мутации у родственника – 2800-3500 тыс. руб.
Если в семье уже имелись подобные случаи генетических нарушений, то настоятельно рекомендуется сделать диагностику перед подготовкой к беременности (стоимость исследования – 8-10 тыс. руб.). Такое исследование является единственным способом профилактики возникновения данной патологии у ребенка.
Морской окунь для беременных и кормящих
Во время беременности употребление в пищу морского окуня настоятельно рекомендуется. При питании мясом морского окуня у беременных улучшается функционирование эндокринной системы, наблюдаются существенные улучшения состояния кожи, а большое количество фосфора и кальция позволяют сохранить здоровье суставов и зубов.
Полезен окунь и для плода – благодаря наличию полиненасыщенных жиров Омега-3 и витамина D у ребенка нормально формируется костная система. А содержащийся в относительно большой концентрации йод позволяет не только поддерживать функционирование щитовидной железы мамы, но и создать предпосылки для формирования нормального иммунитета у ребенка.
Морской окунь при грудном вскармливании также будет полезен, поскольку при этом продолжается снабжение ребенка всеми полезными веществами этой рыбы.
Симптомы и отличия
Диагностика
Только у врача можно определить ее и отличить от других отклонений и нарушений зрения.
Что учитывается при осмотре
- возраст больного — это заболевание более вероятно встретить у человека после 40 лет, хотя в некоторых случаях его проявления хорошо заметны
- его жалобы на отклонения в зрении — зачастую симптомы уже дают основания заподозрить возрастные изменения в глазах
- объективные данные, которые получают на специальном оборудовании — они дают полную картину общего состояния глаз пациента
На осмотре врач смотрит на рефракцию, оценивает объем аккомодации, исследует каждый глаз, чтобы определить ближайшую точку видения для них, производит исследование его структур, состояние сетчатки.
Так как старческая дальнозоркость часто сочетается с глаукомой, врач обязательно проверяет глазное давление, проверяет на наличие пока скрытых для пациента заболеваний глаз. Это поможет подобрать оптимальное лечение, поможет скорректировать зрение и не допустить его необратимой утраты.
Культура
Статья в New York Times от июля 1977 года, которая в последующие недели была перепечатана во многих газетах по всей стране, впервые привлекла внимание многих людей к этой болезни.
Беспорядок был показан в шоу Nip / Tuck в эпизоде » «. В фильме TLC « Born Without a Face» изображена Джулиана Ветмор, которая родилась с самым тяжелым случаем в истории болезни этого синдрома, и у нее на лице отсутствуют 30-40% костей.
В 2010 году документальный фильм BBC Three « Люби меня, люби мое лицо» рассказывал о случае человека, Джоно Ланкастера, с этим заболеванием. В 2011 году BBC Three вернулись к Джоно, чтобы осветить его и его партнершу Лору стремление создать семью в фильме « Что, если мой ребенок родился, как я?». , который впервые вышел в эфир в рамках трех сезонов программ BBC о воспитании детей. Первый фильм был показан на BBC One незадолго до первой трансляции второго фильма на BBC Three. Третий фильм Ланкастера BBC Three «В поисках семьи на Facebook» , посвященный усыновлению , вышел в эфир в 2011 году.
В Wonder , то детский роман , главный герой является ребенок , который имеет синдром Treacher Collins. В ноябре 2017 года вышла экранизация 2017 года с Джулией Робертс , Оуэном Уилсоном и Джейкобом Трембле в главных ролях . Элисон Мидстокк , актриса и модель с этим заболеванием.