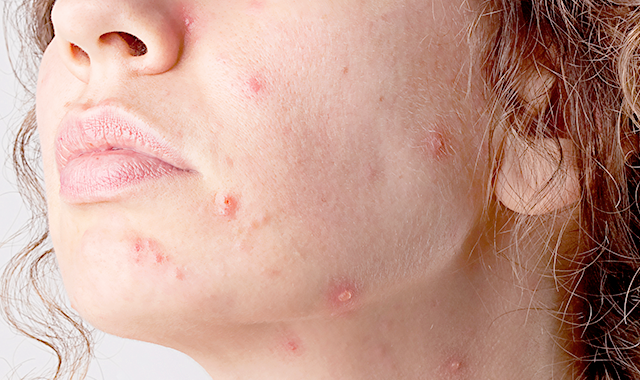
Первичные дефициты белков комплемента
Недостаточность белков комплемента проявляется по-разному в зависимости от того, какой (или какие) белки отсутствуют.
Выделяют три группы заболеваний, связанных с первичным дефицитом комплемента:
- Комплемент-зависимые иммунодефицитные синдромы
- Комплемент-ассоциированные аутоиммунные болезни
- Наследственный ангионевротический отёк Квинке—Ослера.
Комплемент-зависимые иммунодефицитные синдромы
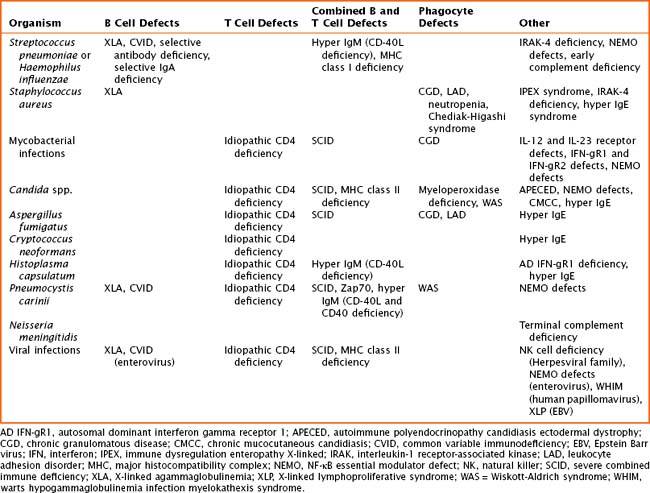
Комплемент-зависимые иммунодефицитные синдромы — заболевания, сопровождающиеся недостаточностью антибактериальной защиты организма. Они проявляются частыми инфекционными процессами в различных органах и тканях. Поскольку белки комплемента при активации играют роль хемоаттрактантов и опсонинов, обеспечивая эффективную функцию фагоцитирующих клеток, то при дефиците компонентов комплемента формируется вторичная недостаточность функции макрофагов и нейтрофильных гранулоцитов. Особенно часто инфекционные процессы при этом вызваны стрептококками, в частности пневмококками, и Haemophilus influenzae. В эту группу включают недостаточность С3b-инактиватора, белков С3, С6 и С8.
Недостаточность С3b-инактиватора. С3b-инактиватор играет роль ингибитора альтернативного пути активации комплемента. При его отсутствии происходит быстрое потребление С3-компонента (вторичный дефицит С3), который в нормальных условиях принимает активное участие в антибактериальной защите организма. Белка С3 у больных в плазме примерно 20 % от нормы. Однако на 75 % он представлен С3b-фрагментом. Уровень нативного С3 составляет всего 5 % от нормы. Скорость расщепления С3 у больных повышена почти в 5 раз. Показано, что через 2 часа после инъекции нативного С3 расщеплению подвергается 40 % введённых молекул. Помимо вторичного дефицита С3 формируется вторичная недостаточность белка С5, однако она менее выражена (примерно 40 % от нормального уровня). Заметно снижена концентрация фактора В — 5 % от нормы (расщепление фактора В происходит под влиянием фактора D). Уровень пропердина снижен незначительно. Больные при этом заболевании страдают различными бактериальными инфекциями.
Недостаточность С3. Недостаточность С3-компонента комплемента также проявляется различными бактериозами. В основе заболевания, в отличие от недостаточности С3b-инактиватора, лежит первичный дефицит С3-белка.
Комплемент-ассоциированные аутоиммунные болезни
Недостаточность белков комплемента провоцирует возникновение аутоиммунных заболеваний, прежде всего (1) красной волчанки, (2) так называемого волчаночно-подобного синдрома и (3) ревматоидного артрита. Часто поражаются почки по типу гломерулонефрита. У больных также описаны пурпура Шёнлейна—Геноха и полимиозит. К этим заболеваниям относятся недостаточность белков С1, С2, С4 и С5. Гены этих белков сцеплены с генами иммунного ответа (генами МНС), поэтому дефекты их, как правило, обоюдны.
Недостаточность С2. Недостаточность С2 является самым частым вариантом первичного дефицита белков комплемента. С2 синтезируют фиксированные и блуждающие макрофаги, фагоцитарная функция которых при этом не нарушена.
Наследственный ангионевротический отёк Квинке—Ослера
К третьей группе состояний, связанных с первичной недостаточностью комплемента, относится наследственный ангионевротический отёк Кви́нке—О́слера, в основе которого лежит недостаточность С1-ингибитора. У отдельных больных при этом возникают аутоиммунные процессы, прежде всего красная волчанка.
Первичные дефициты клеточного иммунитета
К первичным дефицитам клеточного иммунитета относятся следующие заболевания:
- Синдром Ди Джорджи
- Синдром Дункана
- Недостаточность пуриннуклеозидфосфорилазы
- Оротацидурия
- Биотин-зависимые ферментопатии.
Синдром Ди Джорджи
В основе синдрома Ди Джо́рджи (Di George) лежит гипоплазия тимуса. Синдром описан в г. Считается, что это заболевание не является наследственным, оно возникает в результате приобретённого нарушения органогенеза в области III—V жаберных дуг (глоточных карманов) на 6—8 неделе беременности. Поэтому, кроме порока тимуса, отмечаются дефекты околощитовидных желёз, сердца и крупных сосудов, а также орофациальные пороки (микростомия, микрогнатия, гипертелоризм, низкое расположение ушных раковин).
Результатом гипоплазии паращитовидных желёз является дефицит парат-гормона и персистирующая гипокальциемия, вследствие чего развивается судорожный синдром, который может проявиться уже в первые часы жизни (неонатальная тетания). Причиной смерти детей в более старшем возрасте служат осложнения, связанные с пороками развития сердца.
Нарушения, затрагивающие Т-лимфоциты, могут быть как очень глубокими, так и едва заметными. В любом случае функция Т-клеток с возрастом восстанавливается и к 5 годам, если ребёнок остаётся жив, не удаётся обнаружить их недостаточности. Антиген-независимый этап созревания Т-клеток при этом происходит вне тимуса — в многослойных плоских эпителиях, прежде всего в эпидермисе. Одним из эффективных способов лечения синдрома Ди Джорджи является трансплантация эмбриональной ткани тимуса.





Синдром Дункана
Синдром Ду́нкана (Х-сцепленный лимфопролиферативный синдром) — иммунодефицит, характеризующийся повышенной чувствительностью к вирусу Эпштейна—Барр. Ген повышенной чувствительности к вирусу локализован в Х-хромосоме, тип наследования заболевания рецессивный, поэтому болеют мальчики. У больных, перенёсших инфекционный мононуклеоз, развиваются длительное лихорадочное состояние, лимфаденопатия (увеличение лимфатических узлов), лимфоцитоз периферической крови, гепато- и спленомегалия. Позднее формируется В-клеточная лимфома, чаще в терминальных отделах тонкой кишки, от которой больные и погибают. Летальные исходы обусловлены также деструктивным гепатитом, вызываемым вирусом Эпштейна—Барр.
Недостаточность пурин-нуклеозид-фосфорилазы
Недостаточность пурин-нуклеозид-фосфорилазы (ПНФ) наследуется по аутосомно-рецессивному типу. Дети страдают гипопластической анемией и крайне сниженной функцией Т-клеток.
Оротацидурия
Оротацидури́я — наследственное заболевание синтеза пиримидинов, которое проявляется повышенной экскрецией оротовой кислоты (оротата) с мочой, недостаточностью Т-лимфоцитов, мегалобластной анемией и задержкой умственного и физического развития. При этом заболевании снижена активность ферментов оротидил-пирофосфорилазы и оротидил-декарбоксилазы, которые преобразуют оротовую кислоту в нуклеотид-оротидин-монофосфат, необходимый для синтеза нуклеиновых кислот.
Биотин-зависимые ферментопатии
Биотин-зависимые ферментопатии также сопровождаются развитием клеточного иммунодефицита (наследственные дефекты биотинидазы и биотин-зависимых энзимов пируват-карбоксилазы и пропионат-карбоксилазы, участвующих в метаболизме аминокислот с разветвлённой цепью — валина, лейцина, изолейцина). Заболевание проявляется уже в периоде новорождённости эпизодами кетоацидоза, неврологической симптоматикой, алопецией, кожными сыпями и непереносимостью белка (рвота, мальдигестия, дегидратация). В моче содержится большое количество органических кислот. Дети отстают в физическом развитии. Из инфекционных процессов наиболее часто развиваются кандидоз и кератоконъюнктивиты. Биотин даёт хороший терапевтический эффект.
Клиника
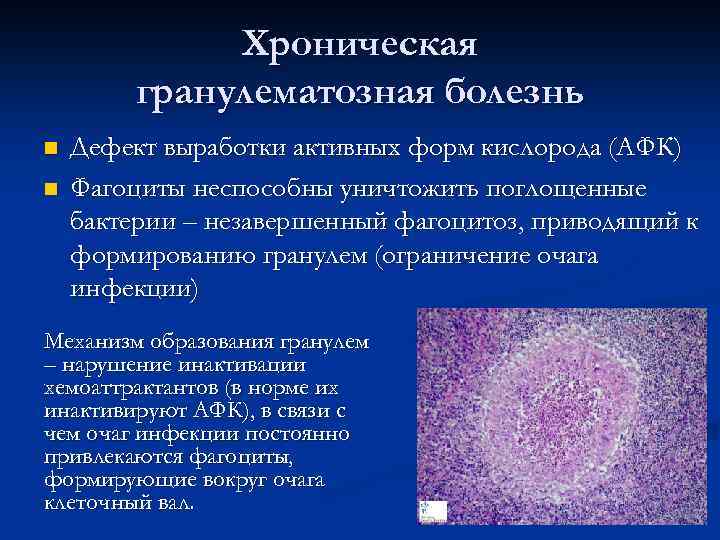
Больные страдают инфекциями в тяжелой форме, которые повторяются периодически. Подвержены инфицированию части тела, которые контактируют с бактериями. Появляются гнойные образования на коже или периодонтит. Хронический гранулематозный периодонтит обуславливается переходом инфекции из оболочки зуба в кость, что касается его корня. Образования на коже проявляется в виде экзем вокруг носа и рта, сопровождающиеся гнойными аденитами. Также в печени человека наблюдаются стафилококковые гнойные воспаления тканей, остеомиелит в костях. В местах попадания в кости и мягкие ткани образуются грамотрицательные бактерии. Если очаг активен и поддерживается микрофлорой, что активно развивается в корне зуба, организм строит барьер для его изоляции, так появляется гранулема. Он изолирует источник инфекции, снижает всасывание и распространение микробов. У больных часто происходит обострение хронического гранулематозного периодонтита, история болезни многих пациентов позволяет говорить о том, что данное заболевание присуще всем, кто имеет гранулематозную болезнь. У больных также наблюдается воспаление лимфоузлов, кишечника, легких и костей. Из грибковых поражений выделяют кандиоз, апергиллез. Часто наблюдается воспаление на месте прививки БЦЖ.
Первичные иммунодефициты
Определение и классификация
Первичные иммунодефициты — это врожденные (генетические или эмбриопатии) дефекты иммунной системы. В зависимости от уровня нарушений и локализации дефекта они бывают:
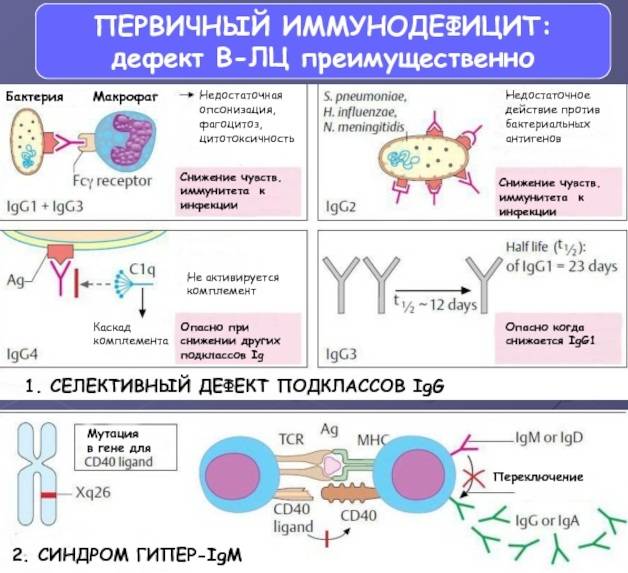
- гуморальные или антительные — с преимущественным поражением системы В-лимфоцитов
- Х-сцепленная агаммаглобулинемия (болезнь Брутона)
- Гипер-IgM синдром
- Х-сцепленная
- аутосомно-рецессивная
- делеция генов тяжелых цепей иммуноглобулинов
- дефицит k-цепей
- селективный дефицит субклассов IgG с или без дефицита IgA
- дефицит антител с нормальным уровнем иммуноглобулинов
- общая вариабельная иммунная недостаточность
- дефицит IgA
- клеточные
- синдром Ди Джоржи
- первичный дефицит CD4 клеток
- дефицит CD7 Т-клеток
- дефицит ИЛ-2
- множественная недостаточность цитокинов
- дефект передачи сигнала
- комбинированные:
- синдром Вискотта-Олдрича
- атаксия-телеангиоэктазия (синдром Луи-Бар)
- тяжелая комбинированная иммунная недостаточность
- Х-сцепленная с полом
- аутосомно-рециссивная
- дефицит аденозиндезаминазы
- дефицит пуриннуклеозидфосфорилазы
- дефицит молекул II класса МНС (синдром лысых лимфоцитов)
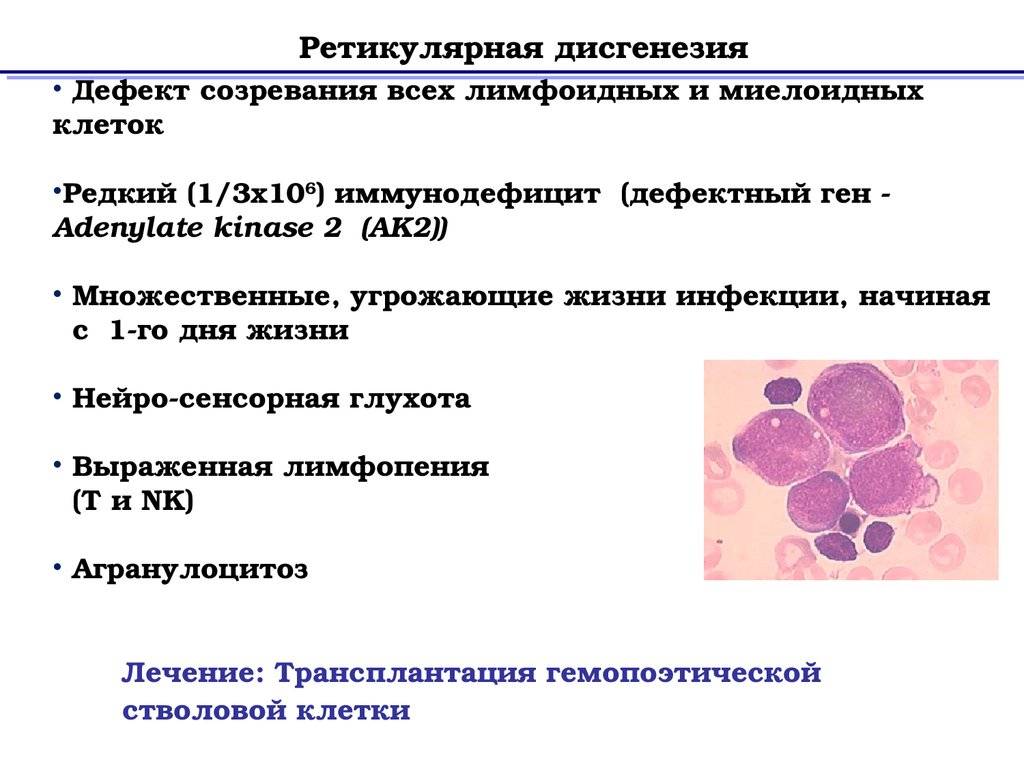
- ретикулярная дизгенезия
- дефицит CD3γ или CD3ε
- дефицит СD8 лимфоцитов
- недостаточность системы комплемента
- дефекты фагоцитоза
- наследственные нейтропении
- инфантильный летальный агранулоцитоз (болезнь Костмана)
- циклическая нейтропения
- семейная доброкачественная нейтропения
- дефекты фагоцитарной функции
- хроническая гранулематозная болезнь
- Х-сцепленная
- аутосомно-рециссивная
- дефицит адгезии лимфоцитов I типа
- дефицит адгезии лейкоцитов 2 типа
- дефицит глюкозо-6-дегидроегназы нейтрофилов
- дефицит миелопероксидазы
- дефицит вторичных гранул
- синдром Швахмана
- наследственные нейтропении
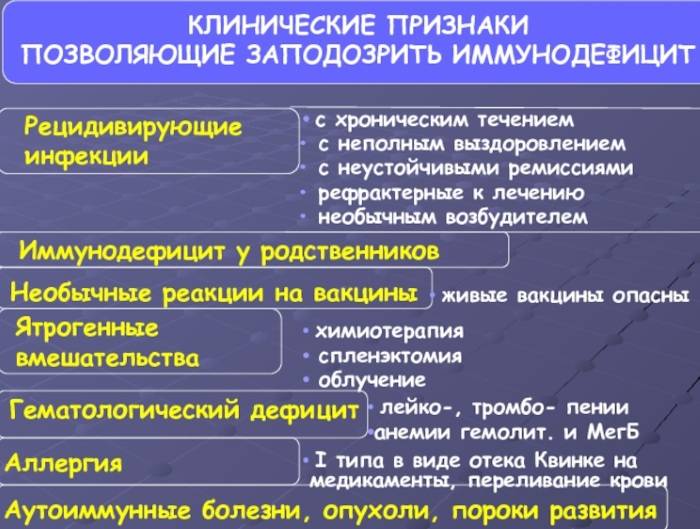
Клиническая картина ИДС
Клиника имеет ряд общих черт:
- 1. Рецидивирующие и хронические инфекции верхних дыхательных путей, придаточных пазух, кожи, слизистых оболочек, желудочно-кишечного тракта, часто вызываемые оппортунистическими бактериями, простейшими, грибами, имеющие тенденцию к генерализации, септицемии и торпидные к обычной терапии.
- 2. Гематологические дефициты: лейкоцитопении, тромбоцитопении, анемии (гемолитические и мегалобластические).
- 3. Аутоиммунные расстройства: СКВ-подобный синдром, артриты, системная склеродермия, хронический активный гепатит, тиреоидит.
- 4. Нередко ИДС сочетается с аллергическими реакциями 1 типа в виде экземы, отека Квинке, аллергическими реакциями на введение лекарственных препаратов, иммуноглобулина, крови.
- 5.Опухоли и лимфопролиферативные заболевания при ИДС встречаются в 1000 раз чаще, чем без ИДС.
- 6. У больных с ИДС часто отмечаются расстройства пищеварения, диарейный синдром и синдром мальабсорбции.
- 7. Больные с ИДС отличаются необычными реакциями на вакцинацию, а применение у них живых вакцин опасно развитием сепсиса.
- 8. Первичные ИДС часто сочетаются с пороками развития, прежде всего, с гипоплазией клеточных элементов хряща и волос. Кардиоваскулярные пороки описаны, главным образом, при синдроме Ди-Джоржи.
Лечение первичных ИДС
Этиотропная терапия заключается в коррекции генетического дефекта методами генной инженерии. Но такой подход является экспериментальным.
Основные усилия при установленном первичном ИДС направлены на:
- профилактику инфекций
- заместительную коррекцию дефектного звена иммунной системы в виде трансплантации костного мозга, замещения иммуноглобулинов, переливания нейтрофилов.
- заместительную терапию ферментами
- терапию цитокинами
- витаминотерапию
- лечение сопутствующих инфекций
- генная терапия
- иммуномодулирующя терапия
В 2018 году российский препарат на основе высокоочищенных прошел . В ходе испытаний была подтверждена безопасность применения лекарственного средства. Планировалось, что после регистрации и завершения дополнительных исследований, препарат возможно будет применять в качестве заместительной и иммуномодулирующей терапии у пациентов со сниженным или отсутствующим уровнем синтеза антител. Средство направлено на обеспечение нормализации уровня иммуноглоублина до оптимальных значений и повышение сопротивляемости организма к патогенам.
Причины возникновения красных пятен
Красные пятна сопровождают заболевания:
- Вирусные заболевания, характеризуются высыпаниями, если человек болеет краснухой, корью. Высыпания по телу, не только на ногах. Повышается температура тела, появляются признаки заболевания. Нельзя исключать сифилис. Если болезнь запущена, красные пятна возникают периодически на ногах. Сыпь яркая, распространяется по телу, прыщики болят. Срочно требуется обратиться за медицинской помощью. Просто устранение сыпи не решает проблемы. Использование народных методов лечения не всегда действенно. Успехов добиваются, используя народные методы переплетая с медикаментозным лечением.
Симптомы краснухи
- Красные прыщики становятся причиной аллергической реакции, у заболевания несколько видов проявлений. Пятно чешется и шелушится – аллерген контактировал с ногами, высыпание появилось на этом участке. Состояние человека остается нормальным, кожа не покрывается пузырьками. Нужно убрать аллерген. Лечение проводится при помощи медицинских препаратов, снимающих отёчность, терапия проводится по назначению врача. Самолечение грозит большими проблемами в дальнейшем.
- Микоз вызывается специальными микробами. Заболевание называется грибком, попадает на ноги из внешней среды. Заразиться можно, сходив в бассейн без обуви – подхватить грибок стопы, начинающий распространяться по телу. Появляются красные пятна, нога сильно чешется, появляются корки, ноготь желтеет и слоится.
Микоз ног
- Пятна на ноге – признак неправильного кровообращения, кровоток нарушается, на коже появляется сыпь. Часто образовываются синяки – первый признак лопнувших капилляров под кожей. Локальные кровотечения бывают мелкими – человек не ощущает дискомфорта, бывают крупными – при нажатии в ногах чувствуется сильная боль.
- Механические и химические раздражители – причины возникновения пятен. Неудобная обувь становится причиной появления красноты на ноге, нога болит, образовывается рана. Если кипящая вода попадает на ногу, образовывается ожог, как и при химическом воздействии.
- Сахарный диабет – причина возникновения сыпи на кожных покровах ног. Кожа на ногах грубеет, становится сухой, начинают появляться язвы. Естественно, лечение направлено на устранение причины. Сахар в крови понизится – пятна сойдут, кожа приобретает естественный цвет, человек избавляется от сопровождающих признаков болезни.
Сахарный диабет
Кроме перечисленных причин, существуют болезни, становящиеся причиной высыпаний на коже ног. Если высыпание болит, стоит обратиться к врачу. Иногда высыпание – единственный симптом серьёзной болезни. Сифилис сложно распознать сразу, высыпание поможет вовремя начать лечение.





Даже мелкие проявления свидетельствуют, что в организме произошёл сбой.
Симптоматика недуга
Часто встречающиеся заболевание у женщин вагинит (кольпит). Это воспалительный процесс, причиной которого является нарушение микрофлоры внутри влагалища в результате размножения инфекционных микроорганизмов. Подострый вагинит наблюдается у 30% женщин.
Нижнее белье стирают, применяя только моющие средства, не вызывающие аллергического раздражения интимных участков тела девочки. Места, пораженные зудом, следует оберегать от расчесывания, в противном случае повышается вероятность занесения новых инфекционных паразитов.
Запущенные формы вагинита увеличивают вероятность возникновения самопроизвольного выкидыша, внематочной беременности, преждевременных родов, преждевременного излития околоплодных вод, внутриутробного инфицирования плода и новорожденного инфекциями, вызвавшими воспаление. Поэтому ведение беременности у женщин с длительно существующим хроническим вагинитом требует особенной тщательности.
Обструктивный бронхит у взрослых
У взрослых обструктивные бронхиты – первично хронические заболевания, которое протекает с прогрессирующей обструкцией дыхательных путей, сопровождающейся нарастающей дыхательной недостаточностью. Хронический обструктивный бронхит относится к хронической обструктивной болезни (ХОБЛ), одной из наиболее тяжелых патологий человека.
Диагностика
Раннее выявление и диагностика имеет важное значение для предотвращения ранней заболеваемости и смерти от системных и легочных инфекций. Диагноз подтверждается аномально низкими уровнями или вообще отсутствующими зрелыми В-лимфоцитами, а также низкой или отсутствующей экспрессией тяжелой цепи м на поверхности лимфоцитов
С другой стороны, уровень Т-лимфоцитов будет повышенным. Окончательный определитель болезни – молекулярный анализ.
Молекулярный анализ также используется для пренатальной диагностики, которая может быть выполнена с помощью отбора проб ворсинок хориона или амниоцентеза, когда мать, как известно, является носителем дефектного гена. Уровни IgG менее 100 мг/дл подтверждают диагноз.
Редко, но диагноз может быть поставлен у взрослых в их втором десятилетии жизни. Это, как полагают, происходит из-за мутации в белке, а не из-за его полного отсутствия.
Лабораторные анализы
На первом этапе необходимо провести количественное измерение IgG, IgM, иммуноглобулина Е (IgE) и иммуноглобулин А (IgA). Уровни IgG следует измерять во-первых, желательно после возраста 6 месяцев, когда уровни материнских IgG начнут снижаться. Во-вторых, уровни IgG ниже 100 мг/дл, как правило, свидетельствует о болезни Брутона. Как правило, IgM и IgA не обнаруживаются.
После того, как уровень антител будет определен как аномально низкий, подтверждение диагноза будет достигнуто с помощью анализа B-лимфоцитных и Т-лимфоцитных маркеров. Уровни CD19 + В-клеток ниже 100 мг/дл. Значения анализа Т-клеток (CD4 + и CD8 +), как правило, увеличиваются.








Дальнейший анализ может быть проведен путем обнаружения ответов IgG к Т-зависимым и Т-независимым антигенам,путем проведения иммунизации, например после введения неконъюгированной 23-валентной пневмококковой вакцины или вакцин от дифтерии, столбняка и H гриппа типа B.
Молекулярно-генетическим исследованием можно установить раннее подтверждение диагноза врожденной агаммаглобулинемии.
Другие обследования
Исследования функций легких занимают центральное место в мониторинге заболеваний легких. Они должны проводиться ежегодно у детей, которые могут выполнять тест (обычно от 5 лет).
Процедуры
Эндоскопия и колоноскопия могут быть использованы для оценки масштабов и прогрессирования воспалительного заболевания кишечника. Бронхоскопия может быть полезной в диагностике и отслеживании хронического заболевания легких и инфекций.
Диагностические мероприятия
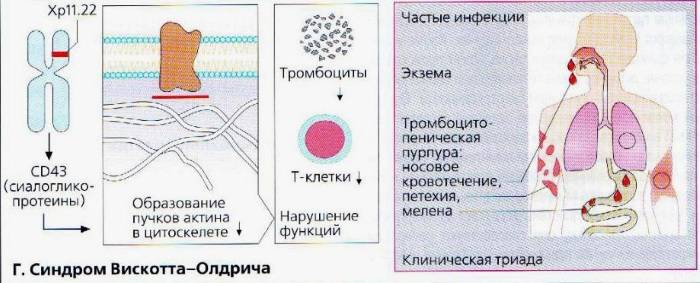
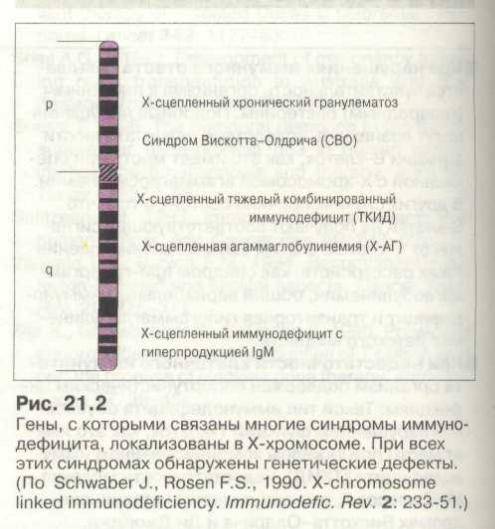
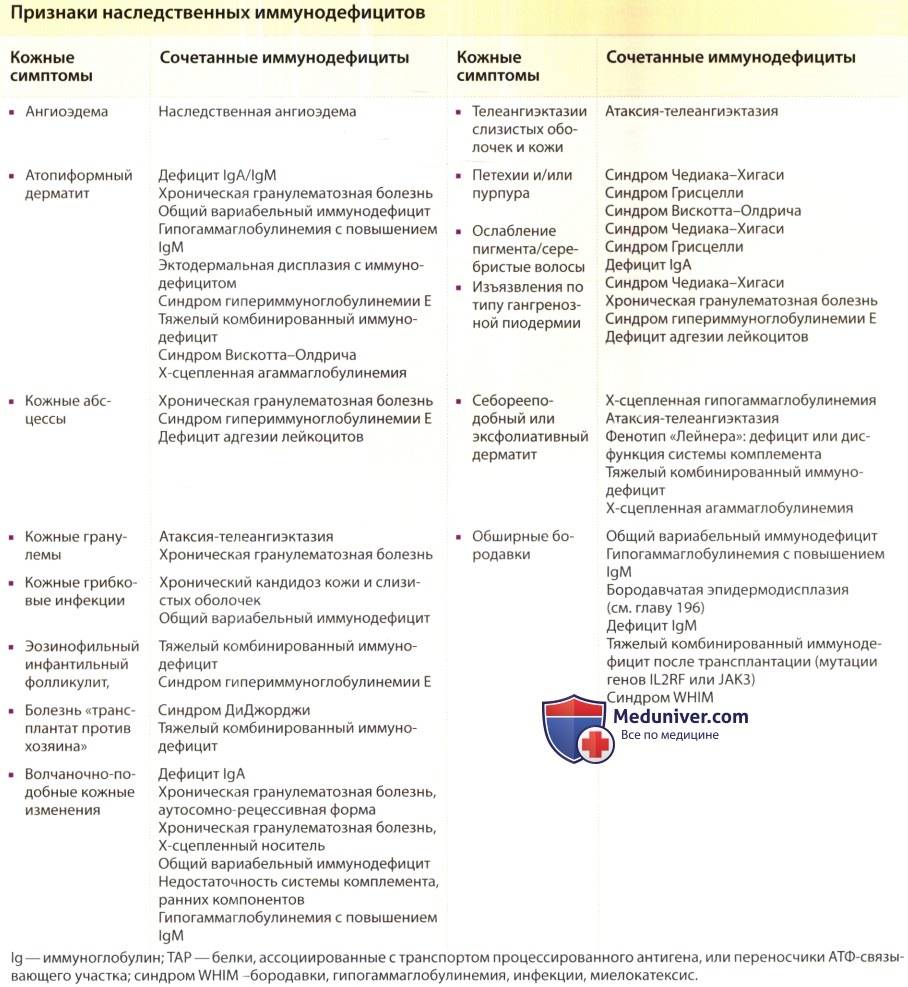
Синдром Вискотта-Олдрича подозревают у всех мальчиков с кровотечениями и врожденной тромбоцитопенией. Признаки острых инфекций и аутоиммунных расстройств могут отсутствовать или быть слабо выраженными.
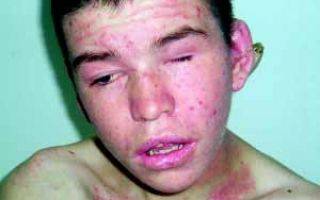
16-летний мальчик с СВО
- Чтобы подтвердить или опровергнуть предполагаемый диагноз, специалисты собирают анализ и выслушивают жалобы больных. Особого внимания заслуживает время возникновения кровотечения, его характер, симптомы инфекционных болезней.
- Поскольку СВО является наследственным заболеванием, очень важен анализ семейного анамнеза. Его выявление у родственников считается важным диагностическим критерием.
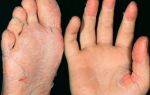
- Затем специалисты переходят к общему осмотру больного, во время которого обнаруживают многочисленные гематомы, петехии и экземы.
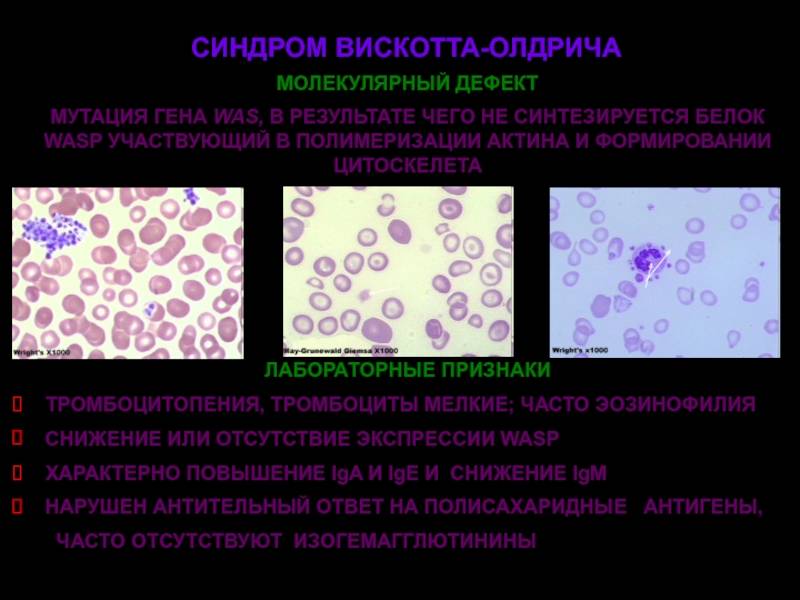
- Данные гемограммы — тромбоцитопения, анемия.
- Иммунограмма — снижение иммуноглобулинов М, повышение иммуноглобулинов A и E, нормальный уровень иммуноглобулинов G.
- В ходе генетического исследования выявляют мутации в гене, кодирующем синтез белка, ответственного за иммунную защиту организма.
- Больным с СВО показана консультация специалистов в области аллергологии, иммунологии, гематологии, медицинской генетики.
Диагностика СВО является сложной и многоуровневой, определяющей дальнейшее лечение больных.
Понятие кератоза и причины его возникновения
Основной причиной кератоза ученые называют постоянное и длительное воздействие солнечной радиации
Кератозом называют заболевание эпидермиса не воспалительного характера, связанное с усиленным ороговением его поверхности и замедленным процессом отшелушивания омертвевших клеток.
Причинами появления кератоза на коже лица могут быть:
- постоянное нахождение под ультрафиолетовыми лучами;
- наследственные (генетические) предрасположенности;
- внешние факторы (с химическим, механическим, лучевым воздействием);
- заболевания инфекционной природы;
- нарушения работы эндокринной и нервной системы;
- венерические болезни;
- злокачественные новообразования органов;
- дерматологические заболевания;
- глубокий авитаминоз.
Основной причиной кератоза ученые называют постоянное и длительное воздействие солнечной радиации на кожную поверхность, которое и приводит к возникновению этой патологии эпидермиса.
В группе риска находятся:
- люди, относящиеся к 1 и 2 фототипу — со светлой кожей, рыжими и светлыми волосами, голубыми или серыми глазами;
- лица с ослабленным иммунитетом;
- носители СПИДа;
- работающие на вредных химических предприятиях или связанные с обслуживанием рентгеновских аппаратов;
- перенесшие трансплантацию внутренних органов.
Первичные иммунодефициты
Определение и классификация
Первичные иммунодефициты — это врожденные (генетические или эмбриопатии) дефекты иммунной системы. В зависимости от уровня нарушений и локализации дефекта они бывают:
- гуморальные или антительные — с преимущественным поражением системы В-лимфоцитов
- Х-сцепленная агаммаглобулинемия (болезнь Брутона)
- Гипер-IgM синдром
- Х-сцепленная
- аутосомно-рецессивная
- делеция генов тяжелых цепей иммуноглобулинов
- дефицит k-цепей
- селективный дефицит субклассов IgG с или без дефицита IgA
- дефицит антител с нормальным уровнем иммуноглобулинов
- общая вариабельная иммунная недостаточность
- дефицит IgA
- клеточные
- синдром Ди Джоржи
- первичный дефицит CD4 клеток
- дефицит CD7 Т-клеток
- дефицит ИЛ-2
- множественная недостаточность цитокинов
- дефект передачи сигнала
- комбинированные:
- синдром Вискотта-Олдрича
- атаксия-телеангиоэктазия (синдром Луи-Бар)
- тяжелая комбинированная иммунная недостаточность
- Х-сцепленная с полом
- аутосомно-рециссивная
- дефицит аденозиндезаминазы
- дефицит пуриннуклеозидфосфорилазы
- дефицит молекул II класса МНС (синдром лысых лимфоцитов)
- ретикулярная дизгенезия
- дефицит CD3γ или CD3ε
- дефицит СD8 лимфоцитов
- недостаточность системы комплемента
- дефекты фагоцитоза
- наследственные нейтропении
- инфантильный летальный агранулоцитоз (болезнь Костмана)
- циклическая нейтропения
- семейная доброкачественная нейтропения
- дефекты фагоцитарной функции
- хроническая гранулематозная болезнь
- Х-сцепленная
- аутосомно-рециссивная
- дефицит адгезии лимфоцитов I типа
- дефицит адгезии лейкоцитов 2 типа
- дефицит глюкозо-6-дегидроегназы нейтрофилов
- дефицит миелопероксидазы
- дефицит вторичных гранул
- синдром Швахмана
- наследственные нейтропении
Клиническая картина ИДС
Клиника имеет ряд общих черт:
- 1. Рецидивирующие и хронические инфекции верхних дыхательных путей, придаточных пазух, кожи, слизистых оболочек, желудочно-кишечного тракта, часто вызываемые оппортунистическими бактериями, простейшими, грибами, имеющие тенденцию к генерализации, септицемии и торпидные к обычной терапии.
- 2. Гематологические дефициты: лейкоцитопении, тромбоцитопении, анемии (гемолитические и мегалобластические).
- 3. Аутоиммунные расстройства: СКВ-подобный синдром, артриты, системная склеродермия, хронический активный гепатит, тиреоидит.
- 4. Нередко ИДС сочетается с аллергическими реакциями 1 типа в виде экземы, отека Квинке, аллергическими реакциями на введение лекарственных препаратов, иммуноглобулина, крови.
- 5.Опухоли и лимфопролиферативные заболевания при ИДС встречаются в 1000 раз чаще, чем без ИДС.
- 6. У больных с ИДС часто отмечаются расстройства пищеварения, диарейный синдром и синдром мальабсорбции.
- 7. Больные с ИДС отличаются необычными реакциями на вакцинацию, а применение у них живых вакцин опасно развитием сепсиса.
- 8. Первичные ИДС часто сочетаются с пороками развития, прежде всего, с гипоплазией клеточных элементов хряща и волос. Кардиоваскулярные пороки описаны, главным образом, при синдроме Ди-Джоржи.
Лечение первичных ИДС
Этиотропная терапия заключается в коррекции генетического дефекта методами генной инженерии. Но такой подход является экспериментальным.
Основные усилия при установленном первичном ИДС направлены на:





- профилактику инфекций
- заместительную коррекцию дефектного звена иммунной системы в виде трансплантации костного мозга, замещения иммуноглобулинов, переливания нейтрофилов.
- заместительную терапию ферментами
- терапию цитокинами
- витаминотерапию
- лечение сопутствующих инфекций
- генная терапия
- иммуномодулирующя терапия
В 2018 году российский препарат на основе высокоочищенных прошел . В ходе испытаний была подтверждена безопасность применения лекарственного средства. Планировалось, что после регистрации и завершения дополнительных исследований, препарат возможно будет применять в качестве заместительной и иммуномодулирующей терапии у пациентов со сниженным или отсутствующим уровнем синтеза антител. Средство направлено на обеспечение нормализации уровня иммуноглоублина до оптимальных значений и повышение сопротивляемости организма к патогенам.
Нормативные правила перемотки
Рукав противопожарного крана в процессе техобслуживания не только перематывается:
- сначала его нужно полностью раскатать;
- затем средство пожаротушения моют и тщательно высушивают;
- только после этого можно проводить гидроиспытания;
- обнаруженные дефекты, подлежащие ремонту, устраняются;
- после этого рукав перекатывается для хранения;
- осуществляется приемка работ заказчиком и заполнение документации.
Во время гидравлических испытаний проверяется работоспособность водопровода, эффективность водоснабжения, уровень водоотдачи и другие факторы.
 Скатка рукава на ручном станке
Скатка рукава на ручном станке
Причины возникновения
Синдром Вискотта-Олдрича у детей определяет мутация единственного гена WAS (Wiskott-Aldrich syndrome) в плече женской Х хромосомы. Он кодирует белок WASp, отвечающий за свертываемость крови и устойчивость иммунитета к инфекции. Wiskott-Aldrich syndrome по длине состоит из двенадцати экзонов, мутация затрагивает G431A, С290Т, G257A участки, отвечающие за синтез белка, приводя к его отсутствию или недостаточности, что, в свою очередь, отражается на цитоскелете клетки. Генетический сбой приводит к развитию тромбоцитопении, атопического дерматита на фоне иммунной недостаточности.
Заболевание СВО, формирующееся в эмбриональный период, характеризуется как первичный иммунодефицит. Состояние обусловлено неспособностью клеток системы обрабатывать информацию о вторжении инфекции и вырабатывать антитела к ней. Всегда сопровождается аномальным изменением формы и количества тромбоцитов. От вида мутации WAS зависит тяжесть клинического течения патологии. Если нарушения вызвали усечение длины белка, сопровождаются более явными и тяжелыми признаками. В случае недостаточного количества синтеза WASp нормального размера клиническая картина значительно мягче.
Методы диагностики
- Сначала проводится осмотр пациента и делается предположение о виде кератомы, определяется размер, локализация новообразования, примерное количество опухолей.
- Далее назначаются дополнительные методы диагностики, чтобы точно установить диагноз.
- Пожилым людям проводится сравнительная диагностика, чтобы обследовать не только кератому, но и другие кожные образования для сравнения.
- Проводится дерматоскопия — осмотр кератомы дерматоскопом, который обеспечивает многократное увеличение.
При вероятности злокачественной динамики нужна гистология. Для этого иссекается небольшая часть опухоли и направляется в лабораторию на анализ.