Диагностика
Поскольку заболевание генетически обусловленное, то основной и единственный способ профилактики врожденных патологий – генетическая консультация при планировании беременности.
Для минимизации рисков, если оба родителя являются носителями мутации, можно провести ЭКО с преимплантационной генетической диагностикой (подтвердить отсутствие заболевания до переноса эмбриона в матку).
Возможна пренатальная диагностика – биопсия ворсин хориона до 12-й недели беременности или амниотической жидкости – после 14-й недели.
При подозрении на СМА у ребенка лучше всего провести генетическое тестирование – выявление мутаций в гене SMN1 и определение числа копий гена SMN2. Это исследование можно считать необходимым и достаточным для подтверждения диагноза СМА. Если оно невозможно или если при тревожных симптомах не выявлено мутаций, то проводят дополнительные исследования:
- анализ крови на уровень креатинкиназы – он позволяет отличить СМА (уровень в норме или незначительно повышен) от мышечной дистрофии Дюшенна (уровень креатинкиназы повышен существенно);
- электронейромиография (ЭНМГ) – инструментальное исследование нервно-мышечной передачи и возбудимости мышц позволяет отличить СМА от бокового амиосклероза и дистрофии Дюшенна;
- биопсия мышечных волокон или икроножного нерва – наиболее информативный анализ для подтверждения диагноза СМА.
Симптомы
 Признаки патологии зависят от ее разновидности и степени тяжести. Атрофия Верднига-Гоффмана может быть выявлена во время внутриутробного развития. В такой ситуации наблюдается низкая двигательная активность плода.
Признаки патологии зависят от ее разновидности и степени тяжести. Атрофия Верднига-Гоффмана может быть выявлена во время внутриутробного развития. В такой ситуации наблюдается низкая двигательная активность плода.
После появления на свет такие дети остаются малоактивными – они практически не двигаются. Мышцы конечностей могут быть поражены изначально. По мере прогрессирования недуга аномальные процессы затрагивают мышечные ткани головы и шеи.
Дети с этим нарушением не способны самостоятельно сидеть. Иногда они все же садятся, но это происходит довольно поздно и только при помощи взрослых. Патология 1 и 2 типа нередко приводит к смерти.
Со стороны заболевание может быть практически незаметно, поскольку дефицит мышц компенсируют жировые и соединительные ткани. При этом у детей с таким нарушением может наблюдаться дрожание рук или подергиваться язык. Отличительной особенностью заболевания считается уплощение грудной клетки. Это обусловлено ослаблением тонуса мышц.
По мере прогрессирования недуга атрофия приводит к потере сухожильных рефлексов, искажению структуры тела и нарушению пропорций. При этом руки напоминают птичьи лапы – локти сгибаются, руки сильно прижимаются к телу, кулаки сжимаются. Эти признаки возникают на поздних стадиях. В данной ситуации речь идет об артрогрипозе, при котором на месте мышц появляются соединительные и жировые ткани.
У взрослых пациентов заболевание имеет медленное развитие. Они могут долгое время совершать простые бытовые действия. Когда заболевание приобретает запущенный характер, возникает потребность в постоянном контроле.
Нередко спинальная атрофия вызывает сопутствующие нарушения. К ним относят систематическое развитие сложных пневмоний и прочих инфекционно-воспалительных патологий.
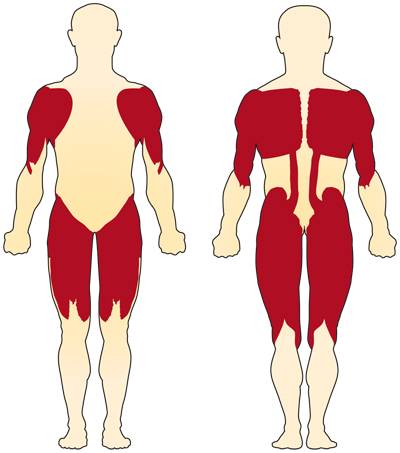
Мышцы, страдающие при спинальной атрофии
Классификация
Самой распространенной формой СМА у детей является проксимальная. Она представлена несколькими видами заболевания, не все из которых становятся очевидными сразу после рождения ребенка.
- Болезнь Вердинга-Гоффмана – СМА 1 типа, тяжелая младенческая болезнь, которая проявляется в первые полгода жизни ребенка. Прогнозы при ней самые неблагоприятные, большинство пациентов погибают. Ребенок с СМА 1 типа не может ни стоять, ни сидеть, ни переворачиваться самостоятельно. У многих новорожденных нарушены сосательные и глотательные рефлексы. Часто отсутствует возможность самостоятельного дыхания или дыхание затруднено.
- Атрофия Дубовица – СМА 2 типа, поздняя младенческая. Проявляется обычно в возрасте от полугода до полутора лет и позднее. Ходить, стоять ребенок не может, но способен сидеть, питание не нарушено, он вполне справляется с задачей глотания, сосания. Сколько проживет малыш, зависит от того, в каком состоянии находятся дыхательные мышцы.
- Атрофия Кугельберга-Веландер – СМА 3 типа, инфантильная. Обычно обнаруживается в возрасте от полутора лет, чаще в два года. Прогностически более благоприятная форма. Маленькие пациенты могут стоять, сидеть, перемещаться, но испытывают сильнейшую слабость, а потому в большинстве случаев нуждаются в инвалидном кресле, без которого нормальная жизнедеятельность для них затруднена.
- Атрофия Кеннеди – СМА 4 типа, бульбоспинальная. Обычно считается взрослой формой, но изредка выявляется и у детей после 15 лет. На длительность жизни влияет редко, ослабление мышц происходит медленно, постепенно, человек, который вел обычную жизнь и считал себя здоровым, со временем становится инвалидом и утрачивает способность перемещаться самостоятельно.
У детей регистрируются не только изолированные формы СМА, когда, кроме дистрофии мышц, ничего не беспокоит, но и сочетанные формы, когда спинальная атрофия – не единственный диагноз и у ребенка есть другие генетические или врожденные проблемы, например, пороки сердца и сосудов, олигофрения.
Болезнь
«Спинальная», потому что большинство двигательных нейронов расположены в спинном мозге. «Мышечная», потому что СМА в основном влияет на мышцы. «Атрофия» — это медицинский термин, обозначающий истощение или уменьшение веса, что обычно происходит с мышцами, когда они неактивны.
СМА влияет на мышцы всего тела, хотя наиболее сильно поражаются мышцы, наиболее близкие к туловищу тела — плечи, бедра и спина. Иногда СМА может повлиять на кормление и глотание. Вовлечение дыхательных мышц (мышц, участвующих в дыхании и кашле) может привести к повышенной склонности к пневмонии и прочим проблемам с легкими.
Интеллект нормален, и часто наблюдается, что люди с СМА веселые и общительные.
СМА является относительно распространенным «редким заболеванием». Приблизительно один из 6000 новорожденных страдает, и примерно каждый 40-й человек является генетическим носителем. Нет никакого лечения, но есть некоторые многообещающие методы лечения, проверяемые в клинических испытаниях.

Лечение
На сегодняшний день не имеется достаточно методов лечения, которые помогли бы избавиться от патологии
Именно поэтому важно отметить то, что выживаемость людей с такой болезнью очень низка. По этой причине наблюдение у ортопедов почти не нужно
Лечение шиной необходимо лишь тогда, когда случаются переломы, что является частым случаем при ослабленном аппарате мышц.
В той ситуации, когда диагностировано заболевание 2 или 3 типа, применяют лечение физической терапией, что необходимо для нормального функционирования суставов. Более радикальное, а точнее, оперативное лечение показано при тяжелой степени поражения суставов.
Лечение медикаментами состоит в следующем:
- Прием лекарств, которые своим действием проводят нервно-мышечные импульсы. Благодаря таким препаратам, улучшается иннервация пораженных мышц, что является причиной для замедления прогрессии патологии.
- Применение лекарств, улучшающих обмен веществ и кровообращения. Такое лечение замедляет прогрессию болезни.
- Использование лекарств, которые активизируют кровоснабжение нервной системы.
Очень важно соблюдение диеты, что, зачастую, не практикуется лечащими врачами. Это необходимо потому, что именно продукты питания помогут восполнить в организме недостаток питательных веществ, витаминов и микроэлементов, которые являются необходимыми при болезни
Стоит отметить, что наследственное заболевание у ребенка нельзя предотвратить никакими методами профилактики. Единственным вариантом можно назвать своевременное обращение к врачу, чтобы как можно раньше начать лечение. В этом случае можно достичь уменьшения масштаба поражений. Если же болезнь – симптом иной патологии, лечение направлено именно на ее устранение.
В любом случае, хоть такую болезнь у ребенка и невозможно вылечить до конца, но при своевременном начале лечения можно облегчить ее течение.
Что такое амиотрофия Верднига-Гоффмана?
Спинальная амиотрофия 1-го типа или, по-другому, спинальная амиотрофия Верднига-Гоффмана
— это особое заболевание нервной системы, передающееся по наследству (чаще всего от обоих родителей). Эта патология характеризуется наличием мышечной слабости практически во всей мышечной системы организма. Ребёнок, страдающий от такого заболевания, не может самостоятельно сидеть, передвигаться и обслуживать себя.
К большому сожалению в мире не существует лекарства от этого типа заболевания. Максимум, что могут предложить врачи в наше время — дородовая диагностика. Такое обследование помогает избежать рождения больного малыша в семье.
Свое название патология получила от двух учёных, впервые описавших её в конце 19 в. В настоящее время под понятием спинальной амиотрофии понимается несколько форм болезни, отличающихся клинически. Но все они при этом связаны одним и тем же генетическим дефектом, которым обладают родители ребёнка.
Клиническая картина заболевания
Спинальная амиотрофия имеет несколько форм и разновидностей
, каждая из которых отличается возрастом появления характерных симптомов, тяжестью протекания заболевания и продолжительностью жизни пациентов.
Обычно эта патология приводит к инвалидности
, поскольку нарушается двигательная система организма, и пациент не способен ни самостоятельно передвигаться, ни самостоятельно себя обслуживать. При тяжелых клинических ситуациях может понадобиться постоянный врачебный контроль в повседневной жизни.
Передвигаться такому больному помогают инвалидные кресла, ходунки, костыли, трости. К смертельному исходу такое заболевание может привести только в том случае, когда появляются осложнения со стороны дыхательной и сердечно-сосудистой системы (при пневмониях и сердечной недостаточностью).
Под воздействие патологии не попадают чувствительные нервные волокна
, поэтому у ребёнка сохраняются все виды чувствительности. Не страдают также интеллект и ментальные функции, так при обучении ребёнок совершенно нормально воспринимает и усваивает информацию.
Классификация заболевания
В зависимости от возраста, при котором появились характерные симптому заболевания, амиотрофия Верднига-Гоффмана делится на несколько видов :
- Врожденная форма патологии . Примерный возраст появления изменений: от 0 до 6 месяцев. Обычно характеризуется слабым внутриутробным шевелением плода. При врождённой форме мышечная гипотония наблюдается с первых дней жизни малыша. В течение короткого времени происходит угасание глубоких рефлексов: ребёнок слабо кричит, плохо сосет молоко матери или соску, не может держать головку. Иногда случается так, что эти симптомы проявляются несколько позже, поэтому малыш может учиться держать головку и сидеть, но, поскольку имеется нарушение, эти навыки у него не разовьются. Также врожденная форма может сопровождаться бульбарными нарушениями, снижением глоточного рефлекса и фасцикулярными подергиваниями языка. Врожденная форма считается наиболее злокачественной и часто может сочетать в себе ещё и олигофрению, деформации грудной клетки, и 4 степени сколиоза . Быстрая обездвиженность и парез дыхательной системы приводит к дыхательной недостаточности и впоследствии к летальному исходу;
- Ранняя детская форма. При данной разновидности патологии первые симптомы могут проявиться после 6 месяцев. К этому моменту дети имеют нормальное физическое и психическое развитие. Они начинают потихоньку приобретать первые естественные навыки, вроде умения держать головку, стоять, садиться и переворачиваться. В большинстве случаев, при наличии данного типа заболевания, дети так и не научатся ходить. На начальной стадии возникают парезы в нижних конечностях, затем довольно быстро они развиваются в верхних конечностях и во всей мускулатуре. Наступает мышечная гипотония, угасают глубокие рефлексы, может проявиться тремор пальцев, непроизвольные мышечные сокращения. На более поздних этапах ко всем симптомам добавляются бульбарные нарушения, дыхательная недостаточность (прогрессирующая). Эта форма заболевание протекает медленнее, чем врожденный тип. Больные могут дожить вплоть до 15 лет;
- Амиотрофия Кугельберга-Веландера. Самая доброкачественная из всех форм спинальной амиотрофии. Симптомы проявляются после 2-х лет, иногда в период между 15-ю и 30-ю годами. При данной форме не встречается психической задержки развития, довольно длительное время пациенты способны двигаться самостоятельно. Многие доживают до глубокой старости на полном самообслуживании.
5 Как проявляется болезнь Дубовица?
Болезнь Дубовица становится заметной после первого полугодия жизни. Атрофия мышц мешает ребенку сидеть и ползать. Такие дети начинают ходить намного позднее. При этом им постоянно требуется помощь.
Нарастание симптоматики в этом случае происходит не так стремительно, как при врожденной СМА. Психическое развитие ребенка проходит нормально. При правильном уходе и постоянном поддерживающем лечении ребенок может прожить более 15 лет.

Ребенок с болезнью Дубовица может самостоятельно себя обслужить, делать простые домашние дела. Такие дети могут учиться в обычной школе, если форма заболевания это позволяет.
При болезни Дубовица сохраняются дыхательный и глотательный рефлексы. Поэтому ребенок может нормально питаться. Такие дети часто страдают от заболеваний верхних дыхательных путей.
Ювенильная форма СМА очень часто проявляется в период полового созревания. В этот момент в организме происходит изменение гормонального фона. Поэтому многие болезни и патологии принимают активную форму.
СМА ювенильного типа развивается медленно. Двигательную активность удается сохранить при помощи специальной гимнастики, физиотерапии и других процедур.
Со временем мускулатура нижних конечностей атрофируется. Больной может передвигаться только в инвалидной коляске. Однако сохраняется определенная степень бытовой самостоятельности.
Согласно медицинской статистике взрослая форма спинальной атрофии мышц наблюдается очень редко по сравнению с другими формами болезни.

Первыми признаками СМА у взрослых людей является:
- слабая динамика шеи и головы;
- дрожание (подергивание) языка;
- затрудненная мимика.
Больной сохраняет двигательную активность. Он может работать, обслуживать себя, существовать в социуме. Если амиотрофия, начавшаяся у подростка или взрослого мужчины, была обнаружена на ранних стадиях, поддерживающее лечение будет наиболее эффективным.
Болезнь Кугельберга-Веландера
Наиболее благоприятный прогноз у тех пациентов, у которых была диагностирована атрофия мышечная спинальная III типа. Она может возникнуть в возрасте от года до 20 лет. Чаще всего первые проявления регистрируют в возрасте 2-7 лет. Первыми страдают проксимальные мышцы таза.
У пациентов возникают трудности при ходьбе, беге, вставании из положения на корточках и необходимости подъема по лестнице. Стоит помнить, что клинические проявления данного заболевания в этой форме схожи с прогрессирующей дистрофией Беккера.
Проксимальные отделы рук и плечевой пояс поражаются лишь спустя несколько лет после первых проявлений болезни. Со временем деформируется грудная клетка, появляется фасцикулярный тремор кистей и неконтролируемое сокращение разных групп мышц. При этом снижаются сухожильные рефлексы и начинают прогрессировать костные деформации. Изменяется грудная клетка, стопы, голеностопные суставы, появляется сколиоз позвоночника.
Причины спинальной мышечной атрофии
К развитию патологии приводит мутация гена, отвечающего за синтез особого белка SMN. Он локализуется на хромосоме 5q. По мере развития недуга происходит отмирание двигательных нейронов спинного мозга. Как следствие, страдают дыхательные и глотательные мышцы. Также атрофируются ткани лица и туловища.
Передача практически всех разновидностей спинальной атрофии происходит по аутосомно-рецессивному типу. Это означает, что носителями аномального гена должны быть оба родителя. Взрослая форма аномалии скреплена с Х-хромосомой. Потому болезни подвергаются исключительно мужчины.
Что ожидается в будущем
Несмотря на прорыв в лечении болезни, разработки препаратов от мышечной атрофии продолжаются. Ученые ищут возможность полностью избавиться от дефектного гена. Для этого ведется разработка следующих направлений генной инженерии:
- коррекция и замещение дефектного гена SMN1;
- усиление активности гена SMN2;
- защита мотонейронов;
- протекция мышц.
Генная инженерия использует технологию внедрения «векторов». Специальный вирусный «вектор» разрабатывают в лаборатории. Он попадает в организм, встраивается в поврежденный фрагмент ДНК.

После этого ген SMN1 активируется и производит необходимый белок. Технология «векторной» инженерии должна быть протестирована для обнаружения долгосрочных побочных эффектов.
Диагноз СМА — это не приговор
Важно не сдаваться, а искать помощь для больного ребенка. Ежедневная забота близких, лечебная физкультура и гимнастика, участие медицинского персонала и врачей помогут справиться с заболеванием
Ольга Гладкая
Автор статей: практикующий врач Гладкая Ольга. В 2010 году окончила Белорусский Государственный Медицинский Университет по специальности лечебное дело. 2013-2014 – курсы усовершенствования «Ведение пациентов с хронической болью в спине». Ведет амбулаторный прием пациентов с неврологической и хирургической патологией.
Лечение
Основная цель исследований, направленных на терапию спинальной мышечной амиотрофии, связана с повышением уровня белка SMN. В настоящее время лекарственные препараты проходят испытания, и официальная российская медицина их не использует.
Лечение сегодня включает лекарства, которые улучшают прохождение нервных импульсов. Назначаются ноотропные препараты, основная задача которых – улучшение работы головного мозга. Назначаются биологически активные добавки, способствующие улучшению обмена веществ. Показана витаминотерапия, в частности, прием витаминов группы Б.
Средства влияющие на нервно-мышечную проводимость:
- Альфа-липоевая кислота
- Ацетил Л-карнитин
- Альфа-глицерофосфохолин
Витамины и витаминные комплексы:
- Тиамин (B-1)
- Пиридоксин (B-6)
- B-комплекс
Важными методами лечения являются массаж, физиотерапия, нейромышечная стимуляция. Назначается ЛФК. Физические упражнения помогают поддержать силу, с другой стороны, выполнение их в обществе, походы в бассейн помогают социализироваться, общаться с другими людьми.
Больным СМА рекомендовано соблюдение диеты. Продукты питания – источник веществ, необходимых мышцам. Так, необходимые аминокислоты содержатся в зерновых, мясе, рыбе, грибах, орехах, кисломолочных продуктах. Рекомендованы блюда из овса и пшеницы, бурого риса.
Естественному поддержанию и росту мышц поможет шпинат, брокколи, сельдь, лук, грейпфрут, арбуз. Для повышения тестостерона мужчинам рекомендуют принимать укроп, пастернак, женьшень, петрушку.
Патогенез
Результатом аберрации SMN-гена является недоразвитие мотонейронов спинного мозга, локализующихся в его передних рогах. Следствием становится недостаточная иннервация мышц, приводящая к их выраженной атрофии с потерей мышечной силы и прогрессирующим угасанием способности совершать активные двигательные акты. Основную опасность представляет слабость мышц грудной клетки, без участия которых невозможны движения, обеспечивающие дыхательную функцию. При этом сенсорная сфера на всем протяжении заболевания остается интактной.
Симптомы
Существует несколько типов СМА. Они отличаются тяжестью симптомов и скоростью их нарастания. Чем раньше проявились признаки заболевания, тем стремительнее оно будет развиваться.
СМА 1 типа (синдром Верднига-Гоффмана)
Первые симптомы проявляются до полугода: ребенок вялый, двигательные навыки не соответствуют возрасту. Если он не держит голову в 2 месяца, следует дополнительно проконсультироваться с неврологом. При СМА 1 типа рано становятся заметны проблемы с сосанием и глотанием: малыш плохо ест, часто пеперхивается.
СМА 2 типа (синдром Дубовица)
До полугода (иногда даже до полутора лет) малыш развивается нормально, а затем происходит остановка или даже откат в развитии (регресс): теряются приобретенные навыки – дети не могут больше стоять, ползать, затем сидеть… Часто родители склонны связывать это с некими внешними факторами (прививка, перенесенное инфекционное заболевание и т.п.), поэтому алгоритм диагностики может быть нарушен. Просто помните, что внезапный регресс – это веский повод пройти генетическую диагностику.
СМА 3 типа (синдром Кугельберга-Веландера)
Болезнь впервые проявляется, когда ребенок, казалось бы, прошел все самые трудные этапы в своем развитии – научился ходить и говорить, есть ложкой и пить из чашки… Эти симптомы могут быть обнаружены в 2 года и позже:
- Ребенок часто спотыкается и шаркает ногами;
- бегает медленнее всех своих сверстников или не бегает вовсе;
- с трудом поднимается по лестнице;
- быстро устает при ходьбе;
- жалуется на одышку;
- часто болеет с осложнениями и плохо откашливается при ОРВИ;
- жалуется на изжогу.
На этом этапе СМА диагностировать труднее всего, поскольку схожие симптомы могут быть связаны со множеством других заболеваний. Зато и развиваются они медленно – настолько, что существенно не влияют на общую продолжительность жизни.
Уход в домашних условиях
Кипрей узколистный — фото, полезные свойства, применение в лечении, рецепты
Спинраза
Симптомы у ребёнка и взрослого
Основным признаком болезни СМА является мышечная вялость, слабость и атрофия. Однако у каждой из форм спинальных амиотрофий существуют свои особенности:
- При заболевании Вердинга — Гоффмана первые симптомы могут быть обнаружены ещё во время беременности на УЗИ осмотре, так как плод очень слабо шевелится. После родов отмечается невозможность ребёнка самостоятельно держать голову, переворачиваться и позднее сидеть. Почти всё время малыш лежит в расслабленной позе на спине, не имея возможности свести ноги и руки. Также отмечаются частые проблемы с кормлением, так как младенец испытывает трудности с глотанием. Дыхание зачастую нарушено из-за атрофии рёберной мускулатуры. Практически 70% детей погибают, не дожив до двух лет. После диагностики выявляется недостаточная сформированность передних рогов спинного мозга. Если пациент доживает до 7–10 лет, то у него нарастает выраженность мышечной атрофии и он погибает от острой сердечной, лёгочной недостаточности или из-за проблем с пищеварением. В редких случаях больные доживают до 30 лет, и то исключительно при более позднем начале проявления симптомов (около 2 лет).
- При втором типе спинальной мышечной атрофии ребёнок зачастую может самостоятельно дышать и глотать пищу. Однако со временем происходит прогрессирование процесса, и в более старшем возрасте дети оказываются прикованными к инвалидным креслам. Обычно родители начинают замечать, что ребёнок часто спотыкается, падает и у него подгибаются колени. Постепенная невозможность самостоятельно проглатывать пищу появляется с возрастом. Также по мере взросления начинает проявляться сильно выраженное искривление позвоночника (сколиоз). Эта форма считается относительно доброкачественной и позволяет пациентам прожить до старости. В некоторых случаях женщины даже могут выносить и родить ребёнка, однако велик шанс передачи болезни по наследству. При правильном уходе и благодаря регулярным занятиям лечебной физкультурой пациенты могут очень долгое время сохранять дееспособность.
- Ювенильная амиотрофия Кюгельберга — Веландера может впервые регистрироваться в возрасте от двух до восемнадцати лет. На самом раннем этапе симптомы могут отсутствовать, ребёнок полноценно развивается. Постепенно начинает появляться слабость в проксимальных отделах тела, чаще всего в плечах и предплечье. В течение многих лет пациент способен самостоятельно передвигаться и обслуживать себя. Часто наблюдаются мышечные подёргивания (фасцикуляции). Основной пик проявления симптомов регистрируется в возрасте от двух до пяти лет, когда ребёнку вдруг становится сложно бегать, вставать с кровати и подниматься по лестнице. Течение болезни относительно доброкачественное, так как пациент может длительно сохранять возможность самостоятельно передвигаться.
- Бульбоспинальная мышечная атрофия Кеннеди — заболевание, сцепленное с полом, передаётся с Х хромосомой и проявляется исключительно у мужчин во взрослом возрасте. Прогрессирует болезнь медленно и начинается со слабости в мышцах бёдер, затем через 10–15 лет постепенно присоединяются бульбарные расстройства (поражения черепных нервов: языкоглоточного, блуждающего и подъязычного). Так как течение заболевания крайне медленное, то важные функции практически не успевают нарушаться и продолжительность жизни сильно не сокращается. Очень часто болезнь сопровождается эндокринными патологиями: атрофией яичек, снижением либидо, сахарным диабетом.
- Дистальная СМА Дюшена — Арана обычно регистрируется в возрасте 18–20 лет. Первыми поражаются кисти рук, затем полностью верхние конечности. В течение длительного времени постепенно наступает атрофия мышц ног. В крайне редких случаях заболевание останавливается на парезе одной из рук.
- Скапуло-перонеальная спинально-мышечная атрофия Вюльпиана впервые дает о себе знать в старшем возрасте (20–40 лет). Проявляется постепенной атрофией мышц плечевого пояса и разгибателей стопы и голени. Прогноз относительно благоприятный, так как, даже спустя 30 лет с момента начала заболевания, у пациента сохраняется возможность передвигаться самостоятельно.
СМА у беременных связана со множеством осложнений. Зачастую самостоятельно родить женщина не может и ей назначают кесарево сечение.
На рентгеновских снимках видно искривление позвоночника и последующее его исправление с помощью операции