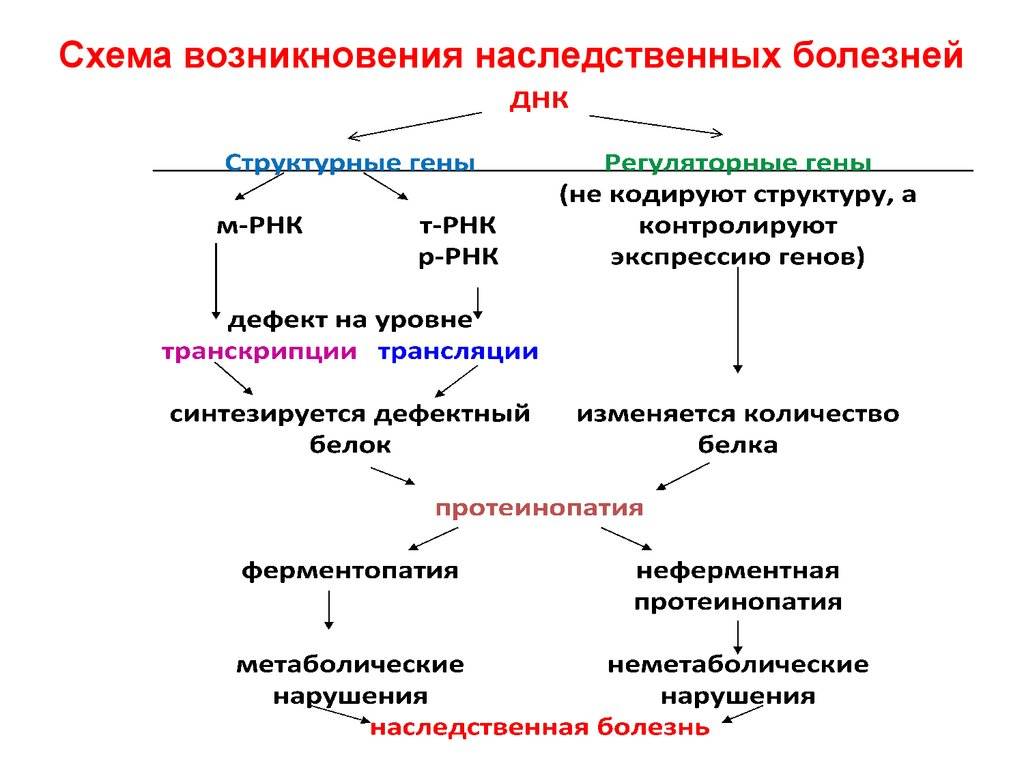
Нейродегенерация в разном возрасте
Дегенеративные болезни нервной системы у детей появляются в раннем детстве. Типичным нейродистрофическим заболеванием является спинальная мышечная атрофия (СМА). Выделяют четыре формы патологии.
Первая появляется в раннем детском возрасте сразу после рождения. Она протекает крайне тяжело. У ребенка отсутствуют рефлексы, он отстает в моторном развитии, плохо питается. При СМА второго и третьего типа симптомы более мягкие. При этом разработано лечение.

Атаксия Фридрейха проявляется на первом десятилетии. Дети уже освоили моторные навыки. На этом фоне четко заметна их прогрессирующая утрата. Появляется шаткость походки, неустойчивость при движении. Вместе с нервной и мышечной тканью, поражается эндокринная система, формируются дегенеративные процессы в позвоночнике. В исходе развивается сахарный диабет, прогрессируют интеллектуальные нарушения. Поражается сердечная мышца.
Нейрометаболические заболевания у детей и подростков
Такие болезни развиваются при поражении метаболизма. Нервная ткань страдает вторично. Типичные нейрометаболические недуги:
- Болезнь Гоше;
- Патология Нимана-Пика;
- Мукополисахаридоз.
Типичным нейрометаболическим синдромом является болезнь Нимана-Пика. При этом «ломается» липидное звено обмена. В клетках откладывается большое количество жирных кислот. При этом увеличиваются печень и селезенка. Дети отстают в развитии. Кроме того, появляется мышечная дистония, нарушение движения.
Бизнес и финансы
БанкиБогатство и благосостояниеКоррупция(Преступность)МаркетингМенеджментИнвестицииЦенные бумагиУправлениеОткрытые акционерные обществаПроектыДокументыЦенные бумаги — контрольЦенные бумаги — оценкиОблигацииДолгиВалютаНедвижимость(Аренда)ПрофессииРаботаТорговляУслугиФинансыСтрахованиеБюджетФинансовые услугиКредитыКомпанииГосударственные предприятияЭкономикаМакроэкономикаМикроэкономикаНалогиАудитМеталлургияНефтьСельское хозяйствоЭнергетикаАрхитектураИнтерьерПолы и перекрытияПроцесс строительстваСтроительные материалыТеплоизоляцияЭкстерьерОрганизация и управление производством
МРТ диагностика
Методы визуализации не являются достоверным методом диагностики. Все дегенеративные нозологии имеют сходные признаки. Но для каждой патологии выделены определенные знаки на МРТ. Поэтому проведение исследования считается обязательным.
«Красные флаги» нейродегенерации:
- Атрофия коры головного мозга;
- Расширение церебральных желудочков.
Специфические изменения также можно диагностировать по МРТ. Например, при болезни Альцгеймера появляется атрофия гиппокампа. Патология Фара отличается отложением кальция в базальных ганглиях. При синдроме Вильсона-Коновалова отмечается усиление сигнала в области бледного шара, базальных ганглиев, черной субстанции. Это типичные места отложения меди.
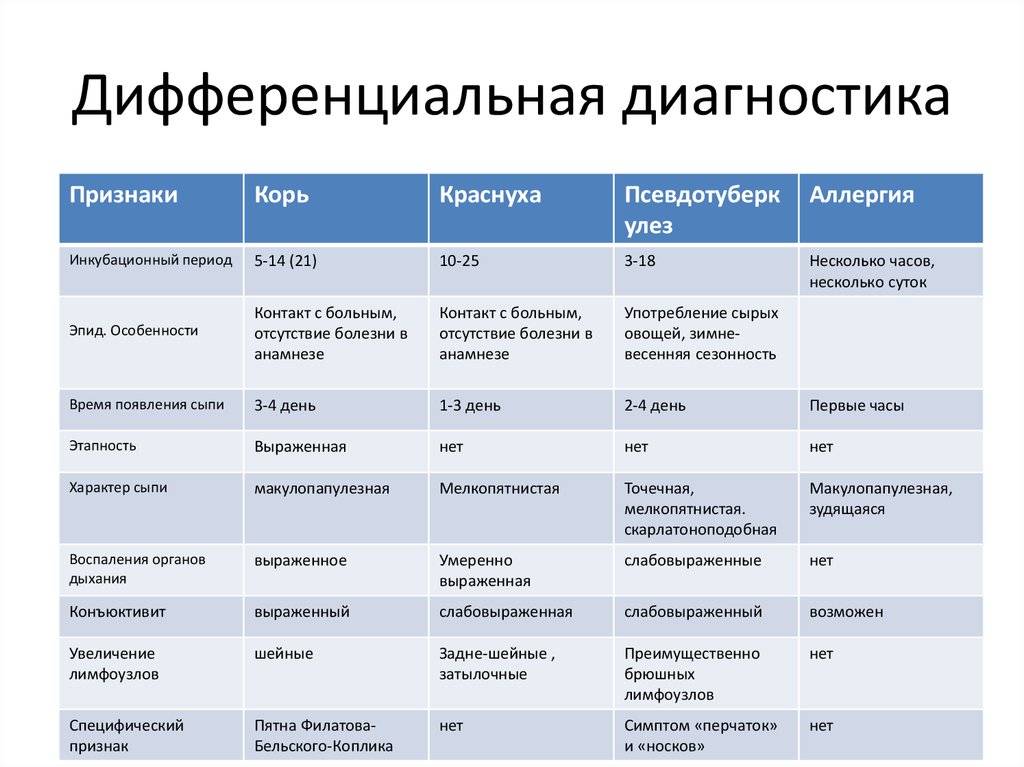
Дифференциальная диагностика дегенеративно-дистрофических заболеваний
Сравнение и дифференциальные признаки некоторых патологий приведем в таблице.
| Область поражения | Патология | Симптомы |
| Кора головного мозга | Болезнь АльцгеймераПатология ПикаФронтотемпоральная деменция | Снижение интеллектаПсихические расстройстваПроблемы контроля настроения и эмоций |
| Подкорковые структуры (базальные ганглии, черная субстанция, полосатое ядро, бледный шар) | Болезнь ФараПаркинсонаДеменция с тельцами Леви | На первый план выходят нарушения двигательных функцийРигидность мышцПостуральная неустойчивость |
| Поражение спинного мозга | Спино-церебеллярная атаксияБоковой амиотрофический склероз | Потеря чувствительности и двигательной функцииПараличи и парезы конечностейФибрилляции |
| Мультифокальное поражение | МСАРассеянный склероз | Клинические признаки появляются в любом отделеВыпада |
Подходы к диагностике и лечению нейродегенеративных патологий
Ранняя диагностика недуга
Основная задача медицины – научиться определять наличие проблемы на латентной стадии. Возможно, при сохранности большего количества нейронов терапия будет иметь успех. Ведь пока почти все патологии считаются неизлечимыми.
Для решения этой задачи ученые предлагают выделять группы риска. Такая стратификация поможет проводить поиск болезни не в огромной популяции, а в небольшой когорте. Например, при болезни Альцгеймера – это e4 вариант аполипопротеина E. При патологии Паркинсона – это контакт с пестицидами.
Второй вариант для раннего поиска – это изучение биомаркеров. Это молекулы, которые позволяют заранее говорить о болезни. При этом их можно обнаружить на здорового человека без симптомов. Биомаркером при болезни Альйгеймера является амилоид. Его большое количество определяют в ликворе еще до появления клиники.
Гипокситерапия
Гипербарическая оксигенация активно использовалась в середине 20 века. Показаний для проведения сеансов в барокамере довольно много. И болезни с деструкцией нейронов не исключение.
Предположительно лечение кислородом стабилизирует свободные радикалы и уменьшает повреждение клеток. Результаты исследований показали, что метод работает. Но эффективность довольно низкая. Кроме того, имеются ограничения при его использовании.
Гипокситерапия основана на вдыхании газовой смеси с пониженным содержанием кислорода. При проведении сеанса пациент поочередно дышит смесью кислорода и обычного воздуха. Метод доступен во многих санаториях, отделениях реабилитации. Масштабных исследований, позволяющих судить о его эффективности, не проводилось.
Моделирование нейродегенеративных болезней
Тестирование лекарств проводится на животных моделях. Обычно используются лабораторные мыши. Ученые способны выводить поколения грызунов с дефектными генами (с патологией Паркинсона, с накоплением бета-амилоидов в клетках). На трансгенных животных тестируют возможности лекарственной терапии. Однако, часто хороший результат в лаборатории не приводит к успеху в клинике.
Некоторые эксперименты проводятся на клеточных культурах. Сейчас наука может длительно поддерживать жизнеспособность клеток. При этом культуры имеют двух- или трехмерное строение. При этом искусственные нейроны приходится длительно выращивать. А погибают они через 2-3 недели. При моделировании на животном возможно отследить весь процесс болезни от первых симптомов до гибели животного.
Способы лечения
Лечение направлено на поддержку мышечных сил, замедление атрофирования.
Цель терапии заключается в том, чтобы пациент как можно дольше передвигался без посторонней помощи, т.к. в постоянном вертикальном положении возникают расстройства дыхания.







Основными способами лечения являются:
- Лечебно-физкультурный комплекс. Это различные активные и пассивные движения. Нельзя подвергать больного усиленной нагрузке. Упражнения должны быть регулярными;
- Ортопедические мероприятия. Применение специальных шин и проведение операций. Эти меры направлены на сохранение самостоятельных передвижений;
- Лекарственные препараты. Медикаменты, поддерживающие обмен веществ, устраняющие дефицит энергии и белка. Больным назначают фосфаден, преднизолон, нифедипин, витамин Е.
Неправильно поставленный предварительный диагноз приводит к ошибкам в диагностике формы НМЗ. Это увеличивает траты на осуществление дорогостоящих анализов и препятствует осуществлению профилактики рецидива, определения генетического статуса предков пациента и подбора адекватного патогенетического лечения, которым можно ограничиться в некоторых случаях.
Полезное видео по теме:
Прогрессирующие наследственные болезни нервной системы
В группу прогрессирующих миопатий (миодистрофий) входят несколько десятков наследственных заболеваний нервной системы, ведущим проявлением которых является прогрессирующее поражение мышечной ткани. Причина заключается в нарушении синтеза ферментов, участвующих в воспроизводстве белков мышечной ткани или двигательных нервных клеток спинного мозга. Болезнь начинается в детском, подростковом, реже — в юношеском возрасте. Заболеваемость составляет до трех случаев на 100 тысяч населения.
Клиническая картина наследственных поражений нервной системы характеризуется нарастающей атрофией мышц, постепенно приводящей к нарушению движений, вплоть до полной обездвиженности. Различают несколько форм миопатии, отличающихся клиническими и генетическими особенностями.
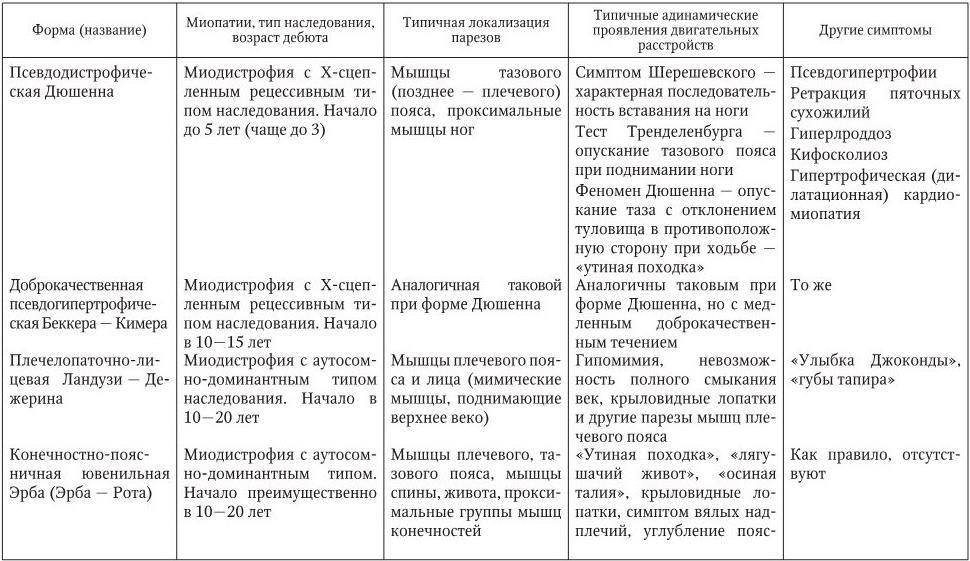
Ювенильная (юношеская) форма Эрба—Рота. Болезнь начинается в возрасте 11-20 лет, болеют чаще мальчики.
Обычно атрофия мышц начинается в проксимальных отделах ног, затем — тазового пояса, туловища и верхних конечностей. При этом тонус и сила мышц снижаются, походка больного становится «утиной», раскачивающейся, усиливается поясничный лордоз, появляется так называемая «осиная талия», лопатки начинают выступать («крыловидные» лопатки), определяется симптом «свободных надплечий». Если атрофируются мимические мышцы, возникает лицо миопата, для которого характерны гладкий лоб, слабость круговых мышц глаз, утолщенные губы и поперечная улыбка.
Нарастающая слабость приводит к тому, что больные не могут встать из положения лежа или сидя и «взбираются» по себе с помощью рук как по лестнице. Диффузно поражаются не только поперечно-полосатые, но и гладкие мышцы, вследствие чего обнаруживаются признаки дистрофии миокарда, вялой перистальтики кишечника. Болезнь медленно прогрессирует, обездвиженность наступает через 15-20 лет.
Псевдогипертрофическая форма Дюшена. Наиболее тяжелая форма миопатии. Наследуется по рецессивному типу, сцепленному с полом. Болеют мальчики.
Болезнь начинается рано, в возрасте около трех лет, и быстро прогрессирует. Первым симптомом является нарушение походки. Атрофический процесс начинается с мышц проксимальных отделов ног и тазового пояса, а затем атрофируются мышцы проксимальных отделов рук, исчезают коленные рефлексы. Очень характерна резкая псевдогипертрофия мышц, особенно икроножных. В терминальных стадиях процесс распространяется на мышцы лица, глотки. К 14-15 годам больные обычно полностью обездвижены; у них возникают заболевания легких, что часто становится причиной смерти.
Плече-лопаточно-лицевая миопатия Ландузи-Дежерина. Относительно доброкачественное заболевание. Начинается в возрасте 20-23 лет и проявляется гипомимией, сглаженностью носогубных складок, недостаточным смыканием век, а также амиотрофией и парезом мышц плечевого пояса, ограничением движений в плечевых суставах, «крыловидными» лопатками. Болезнь прогрессирует медленно, работоспособность сохраняется длительное время.
Помимо первичных мышечных дистрофий (миопатий), существуют вторичные прогрессирующие мышечные дистрофии — амиотрофии. К ним относится невральная амиотрофия Шарко-Мари. Она наследуется по аутосомно-доминантному типу, чаще болеют мужчины. Болезнь начинается около 20 лет. Появляется слабость сначала в дистальных отделах ног, затем — рук, которая сочетается с их атрофией и расстройствами чувствительности. Клинически болезнь напоминает полиневрит, но отсутствуют признаки инфекции или интоксикации.
Лечение проводят анаболическими гормонами (неробол, ретаболил, метилтестостерон):
Витаминами (Е, С, группы В),
Биостимуляторами (АТФ, прозерин, галантамин, дибазол):
Показаны тепловые физиопроцедуры, радоновые, хвойные, сероводородные ванны, массаж.
Профилактика заключается в медико-генетическом консультировании супружеских пар, имеющих больного ребенка; или больных, которые хотят иметь детей.
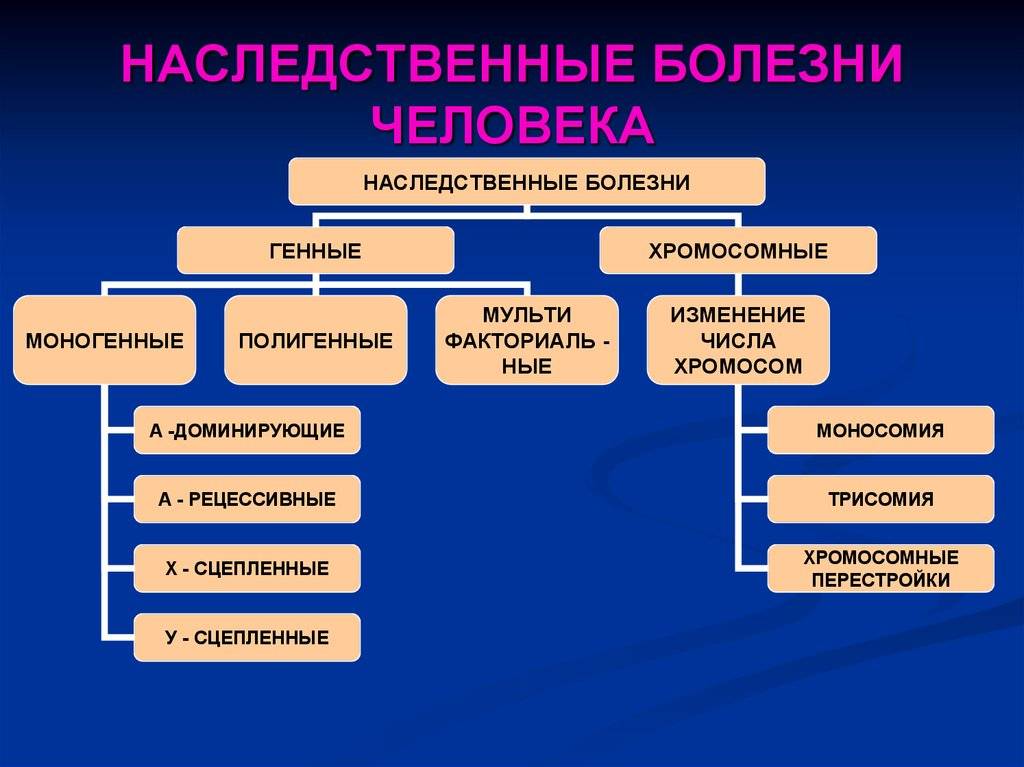
Классификация
Эти патологии можно разделить на несколько классов.
| Атрофии, поражающие центральную нервную систему | G10-G14 |
| Экстрапирамидные поражения с двигательными нарушениями | G20-G26 |
| Другие дегенеративные заболевания центральной нервной системы | G30-G32 |
Дегенерация головного мозга подразделяются по виду патологического белка.
- Таупатии (болезнь Альцгеймера, Пика, парез взора, фронтотемпоральная деменция);
- Синуклеопатия (болезнь Паркинсона, с тельцами Леви, МСА);
- Тринуклеотидная заболевания (Хорея, СМА, атаксия Фридрейха).
Дегенеративно-дистрофический тип
Этот вид патологии возникает при разрушении нервных клеток головного или спинного мозга. К дегенеративным заболеваниям относят болезнь Альцгеймера, Паркинсона, боковой амиотрофический склероз. При этих состояниях идет медленная гибель клеток нервной системы. Ее могут инициировать нарушения ферментных систем или отложения патологических белков в клетке.










Лечения этой проблемы пока не разработано. К сожалению, новые препараты не показали эффективности в устранении причин заболевания. Этиология до сих пор не изучена. Продолжаются исследования в области воспалительной и инфекционной теории их возникновения.
Наследственно дегенеративные заболевания
Этот вид патологии наследуется . Существует аутосомно-рецессивный и доминантный тип наследования. А также некоторые болезни передаются по пути, сцепленному с полом. Типичные примеры наследственных болезней – это мультисистемная атрофия, атаксия Фридрейха, хорея Хантингтона.
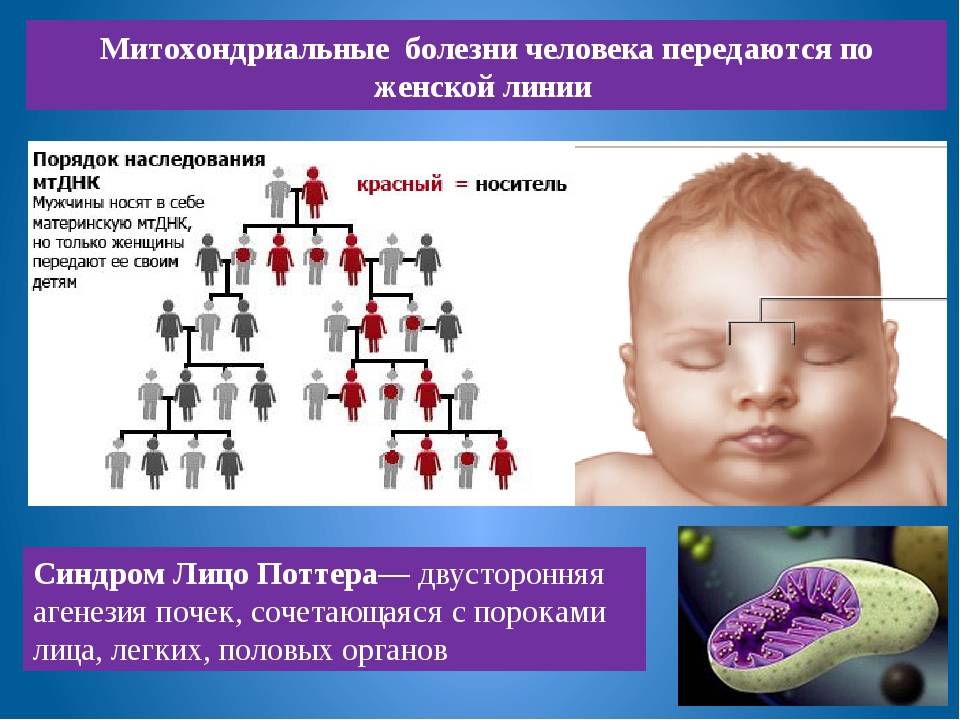
Аутосомно-рецессивный тип наследования характеризуется передачей через несколько поколений. При этом болезнь не проявляется у родителей, а дети могут иметь симптомы заболевания. При наличии дефектного гена у обоих родителей только в 25% случаев возникает недуг. При доминантном наследовании у 50% потомков появляются симптомы заболевания. Патология, сцепленная с полом, передается чаще всего по женской линии.
Нейрометаболический тип недуга
Такой вид патологии появляется при нарушении ферментных систем. Проблемы гомеостаза: отложения протеина, липидов или металлов — вызывают нарушения в нервной ткани.
К наследственным нейрометаболическим болезням юношеского и взрослого возраста относят следующие типы заболеваний:
- Болезнь «кленового сиропа»;
- Митохондриальные патологии;
- Лизосомные болезни накопления (Нимана-Пика);
- Нарушения обмена металлов;
- Болезнь Александера.
Воспалительно-дегенеративные проблемы
Воспаление нервной ткани развивается не так быстро, как в другом органе. Любой нейрон защищен специальной системой. Она носит название гематоэнцефалический барьер. Только грубые поломки в организме дают возможность воспалиться нервному окончанию. Это серьезные нарушения иммунитета или последствия травм. Примером воспалительного недуга является рассеянный склероз.
Аутоиммунные поражения
Типичным примером аутоиммунной патологии является рассеянный склероз. При нем собственные клетки атакуют нервную ткань. Агрессоры проходят гемоэнцефалический барьер и вызывают очаговое поражение клеток.
Аутоиммунные болезни можно контролировать. И даже патологию, которая поражает нервные ткани, научились лечить. Однако, применяются «тяжелые» лекарственные средства.
Сосудистые заболевания ЦНС
Сосудистые заболевания центральной нервной системы – группа болезней, обусловленных патологическими изменениями кровообращения мозга, потерей эластичности сосудов и патологиями работы сердца.
Согласно международной медицинской классификации, они подразделяются на три большие группы: острые нарушения кровообращения головного мозга, хронические нарушения кровообращения и врожденные аномалии сосудов.
Часто развиваются на фоне общих недугов: гипертонии, ревматизма, атеросклероза, дегенеративно-дистрофических поражений шейного отдела позвоночника, диабета и ряда других болезней кровеносной системы. В статистике общей смертности занимают более 21% в раннем восстановительном и отдаленном реабилитационном периоде.
Острые нарушения кровообращения
Возникают на фоне сбоя нормального кровотока в мозге: деформаций, закупорки или полного разрыва сосудов.
Приводят к ишемическим (с вероятностью более 85%) или геморрагическим инсультам, включая субарахноидальные кровоизлияния.
На ранних стадиях развития патология проявляется после интенсивных физических нагрузок или сильного стресса. Затем жалобы на общее недомогание становятся более стойкими, не зависящими от внешних негативных факторов. Качество жизни пациентов ухудшается вплоть до полного исключения возможности продолжения трудовой деятельности
Болезнь сопровождается частыми, резкими скачками артериального давления, сильными головными болями острого режущего или давящего характера, слабостью, онемением в пальцах, ухудшением зрения, нарушениями речи и обмороками.
Хронические нарушения кровообращения
Возникают, как правило, на фоне артериальных гипертензий, атеросклерозов или сочетания этих патологий.
Характеризуются различными морфологическими изменениями сосудов головного мозга, расстройствами центральной гемодинамики и ухудшениями свертывания крови.
На ранних стадиях проявляют себя только общими симптомами недомогания, возникающими после сильного физического и эмоционального переутомления или под воздействием неблагоприятных экологических факторов: могут наблюдаться слабость, расстройства сна и аппетита, значительное снижение объема памяти, затруднение переключения с одного рода деятельности на другой, ухудшение концентрации внимания и темпа умственной деятельности. По мере развития недуга появляются двигательные расстройства: афазии, апраксии, синдром паркинсонизма и прочие нарушения подобного рода.
Врожденные аномалии сосудов
Подразделяются на группу артериальных аневризмов и группу артериовенозных мальформаций.
Артериальные аневризмы развиваются на фоне генетически обусловленного изменения внешнего вида сосудов головного мозга: диффузным расширением просвета артерий, выпячиванием их стенок или прочими деформациями. Распространенность заболевания не высока. Составляет в среднем 1-5% случаев в популяции населения планеты. Тип наследования и этиология заболевания доподлинно не известны. Прослеживается связь с аутосомно-доминантной поликистозной болезнью почек, синдромом Элерса-Данлоса, старческим нейрофиброматозом и синдромом Марфана.






Любая аневризма протекает, как правило, практически бессимптомно. Однако в случае разрыва сосудов может привести к летальному исходу. У детей дошкольного возраста частота разрывов аневризм составляет около 2% случаев, у школьников и подростков около 1%, а у людей в зрелом и пожилом возрасте в пределах 6-10%.
Артериовенозные мальформации, в свою очередь, подразделяются на капиллярные (телеангиэктазии), кавернозные, венозные и артериовенозные. Возникают на фоне утраты связей капилляров между артериальной и венозной циркуляцией. Средняя частота встречаемости составляет девятнадцать случаев на сто тысяч населения. В 60% из них диагностируется в возрасте до тридцати лет.
Характерная особенность данной патологии заключается в том, что ее клинические проявления напрямую зависят от возраста больного. У новорожденных и грудничков ведущими проявлениями болезни являются гидроцефалия и сердечная недостаточность. У детей постарше и взрослых наблюдаются симптомы общего недомогания: головные боли, прогрессирующие психические расстройства, шум в голове и обморочные состояния. У пожилых людей могут начаться частые судороги, напоминающие эпилептические припадки.
Наследственные нервно-мышечные заболевания
Наследственные нервно-мышечные болезни представлены самой обширной группой патологий.
Нервно-мышечные дистрофии представлены 5 большими группами заболеваний, каждая из которых включает множество различных нозологических форм.
К 1 группе нервно-мышечной дистрофии относятся спинальные амиотрофии. К распространенным формам спинальной амиотрофии относятся:
- прогрессирующая спинальная амиотрофия Арана-Дюшенна, наблюдающаяся у взрослых;
- юношеская амиотрофия Кугельберга-Веландера;
- детская форма амиотрофии Верднига-Гоффмана.
Вторая группа нервно-мышечной дистрофии представлена невральными амиотрофиями. Эти заболевания могут быть как семейными, так и спорадическими. Эта группа заболеваний, как правило, проявляется в детском или юношеском возрасте. К этой группе нервно-мышечных дистрофий относятся:
- синдром Русси-Леви;
- невральная форма амиотрофии Шарко-Мари-Тута;
- болезнь Рефсума;
- гипертрофическая интерстициальная невропатия Дежерина-Сотта.
Обширная группа нервно-мышечных заболеваний представлена первичными прогрессирующими мышечными дистрофиями. Проявления первичных прогрессирующих мышечных дистрофий могут быть разнообразными. Яркими представителями этой группы заболеваний являются:
- псевдогипертрофическая детская дистрофия Дюшенна;
- благоприятно текущая псевдогипертрофическая дистрофия Беккера-Кинера;
- ювенальная и конечно-поясничная дистрофия Эрба;
- дистальная мышечная дистрофия;
- окулофарингеальная и окулярная форма дистрофии;
- синдром ригидного позвоночника;
- непрогрессирующая мышечная дистрофия.
К 4 группе нервно-мышечных дистрофий относятся миотонии. Разные фенотипы миотоний наследуются по аутосомно-доминантному пути и характеризуются замедленной релаксацией мышц. К распространенным вариантам наследственных миотоний относятся:
- наследственная парамиотония Эйленбурга;
- миотония Томсена;
- нейромиотония;
- дистрофическая миотония.
К 5 группе нервно-мышечных дистрофий относятся миоплегические синдромы и пароксизмальные миоплегии. К самым распространенным миоплегиям относятся:
- нормокалиемическая миоплегия;
- болезнь Гамсторп;
- гипокалиемическая пароксизмальная миоплегия;
- вторичные варианты пароксизмальной миоплегии.
Все эти заболеваниям имеют свои особенности течения и развития. Некоторые из них позволяют больным жить полноценной жизнью и не испытывать явного дискомфорта, в то время как другие приводят к значительным нарушениям работы мышечного аппарата.
Врожденные поражения нервной системы: причины и лечение патологий
Врожденных нервных заболеваний множество, они затрагивают различные системы и органы. Причины, вызывающие пороки развития, многообразны: воздействие химических веществ (медикаменты, предметы бытовой химии); физических факторов (радиоактивное, ультрафиолетовое излучение, температура); биологических агентов, чаще вирусов. Характер отрицательного влияния перечисленных факторов зависит от периода беременности, интенсивности воздействия и концентрации.
Сирингомиелия — хроническое прогрессирующее заболевание, характеризующееся разрастанием соединительной ткани и образованием полостей в сером веществе головного и спинного мозга.
Основной причиной возникновения сирингомиелии считается дефект эмбрионального развития мозга, а провоцирующими факторами являются травмы, инфекции, тяжелый физический труд. В процессе формирования ЦНС возникают полости в спинномозговом канале и IV желудочке. Кроме дефектов нервной системы у больных сирингомиелией выявляются пороки развития других органов и систем.
Клиническая картина этого врожденного порока нервной системы складывается из четырех групп симптомов: расстройств чувствительности, двигательных нарушений, нарушений вегетативной регуляции, пороков развития других органов и систем.
Расстройства чувствительности проявляются преимущественно снижением болевой и температурной чувствительности по сегментарному типу. Из-за снижения температурной чувствительности больные получают ожоги, с чем чаще всего впервые обращаются к врачу.
Двигательные нарушения при врожденных поражениях нервной системы представлены периферическими и центральными парезами, при поражении продолговатого мозга — расстройствами речи и глотания.
Вегетативные нарушения наблюдаются в виде ожирения, трофических язв на коже, разрушения суставов (артропатия), бледности, синюшности кожи, непереносимости ультрафиолетовых лучей.
Выявляются разнообразные пороки развития: «заячья губа», «волчья пасть», уменьшение или увеличение количества пальцев на конечностях, их срастание, пороки развития сердца, легких и т.д.
Диагностика врожденных пороков развития нервной системы основывается на специфической клинической картине и данных компьютерной томографии (на томограммах видны сирингомиелитические полости или очаги разрастания глиозной ткани).
Лечение всех форм болезни — хирургическое (декомпрессия задней черепной ямки при аномалии Киари, удаление опухоли или дренирование полости). Также проводят симптоматическое лечение, направленное на улучшение обменных процессов в нервной системе (витамины, ноотропные препараты), на улучшение проводимости нервных импульсов при вялых парезах (прозерин, дибазол):
Бальнеолечение (сероводородные, радоновые ванны). При глиозных формах положительный эффект дает рентгенотерапия; при наличии больших полостей, нарушении оттока ликвора проводятся нейрохирургические операции.








При уходе за больными, проведении физиопроцедур необходимо помнить об опасности ожогов и других повреждений вследствие нарушения кожной температуры и болевой чувствительности.
Больным сирингомиелией противопоказаны работа у горячих источников и тяжелый физический труд.
Крамиовертебральные аномалии — врожденные или приобретенные дефекты развития черепно-позвоночного перехода, которые могут сопровождаться поражением головного или спинного мозга (аномалия Киари) или костных структур основания черепа и двух верхних шейных позвонков (платубазия, ассимиляция атланта). При этой врожденной патологии нервной системы могут вдавливаться нижние отделы ствола мозга, мозжечок, каудальные черепные нервы, позвоночные артерии, шейный отдел спинного мозга и его корешки.
Клинически у больных наблюдаются боли в шейно-затылочной области, внезапная мышечная гипотония, нистагм, обморок, апноэ во сне. Диагноз подтверждается данными рентгенографии, миелографии, КТ, МРТ.
Системные дистрофии
Дистрофические процессы в нервной ткани развиваются, как и при миодистрофиях, из-за нарушения синтеза ферментов, участвующих в обмене белков нервных клеток. Второй причиной системных дистрофий является недостаток или избыток веществ, поступающих в мозг из других органов с нарушенной метаболической функцией.
Гепатоцеребральная дистрофия (болезнь Вильсона-Коновалова). Это тяжелое прогрессирующее заболевание, при котором сочетаются поражения головного мозга (подкорковые ядра) и печени. В его основе лежит нарушение синтеза в печени медесодержащего белка церуллоплазмина. Это приводит к увеличению в крови меди, не связанной с церуллоплазмином, отложение ее в избыточном количестве в печени, почках, мозгу, роговице глаза. Встречается с частотой 1 случай на 200 тысяч населения, передается по аутосомно-рецессивному типу.
Болезнь начинается в 10-30 лет. Клинически проявляется симптомами поражения экстрапирамидной системы — ригидностью мышц, приводящей больных к полной обездвиженности, либо крупноразмашистыми гиперкинезами, начинающимися с рук. Наблюдается также поражение печени (циррозы, гепатиты). В течении болезни выделяют две стадии: преневрологическую и неврологическую. Прогрессивно снижается интеллект больных.
Лечение направлено на ограничение поступления меди в организм и усиленное выведение ее из организма. Первое достигается диетой с исключением орехов, шоколада, грибов, какао, виноградных вин, печени трески, бобов; второе — назначением медевыводящих препаратов, к которым относится пеницилламин (купренил).
Назначается по 0,15 г после еды до 2 г в сутки, принимается на протяжении всей жизни.
Для улучшения функции гепатоцитов дают эссенциале, ЛИВ-52, легалон:
Для стимуляции процессов окисления в печени — флумецинол.
Для улучшения выведения желчи — бускопан, феникаберан, но-шпа:
Профилактика заключается в проведении повторных курсов витаминотерапии, особенно В6, С, соблюдении режима труда и отдыха.
Прогноз: до появления эффективных средств летальный исход наступал через 3-5 лет, в настоящее время у 95% больных прогноз благоприятный.
Хорея Гентингтона. Хроническое прогрессирующее заболевание, в основе которого лежит атрофия подкорковых ядер и полушарий большого мозга. Наследуется по аутосомно-доминантному типу.
Возникает у людей старше 35 лет. Проявляется хореическими гиперкинезами, что выражается гримасничанием, причмокиванием, приплясыванием, растопыриванием пальцев рук и ног, вычурными и неожиданными движениями
Наряду с гиперкинезами постепенно ослабляется внимание, ухудшается память, снижается интеллект
Лечение проводят трифтазином, галоперидолом, в сочетании с седативными и общеукрепляющими средствами:
Прогноз для жизни неблагоприятный.
15.2. Прогрессирующие мышечные дистрофии (первичные миопатии)
Прогрессирующие
мышечные дистрофии (ПМД), или
первичные миопатии, характеризуются
дегенеративными изменениями в мышечной
ткани.
Патоморфологические
изменения при
ПМД характеризуются истончением мышц,
заменой их жировой и соединительной
тканью. В саркоплазме выявляются очаги
фокального некроза, ядра мышечных
волокон располагаются цепочками,
мышечные волокна теряют поперечную
исчерченность.








Вопросы
патогенеза остаются до настоящего
времени неразрешенными. В основе
миопатии лежит дефект мембраны мышечных
клеток. Большие надежды возлагаются
на молекулярную генетику.
Различные
формы миопатии отличаются типом
наследования, сроками начала процесса,
характером и быстротой его течения и
топографией мышечных атрофии.
Миопатии
клинически характеризуются слабостью
и атрофией мышц. Существуют различные
формы ПМД.
Наследственные пароксизмальные миоплегии
Пароксизмальные миоплегии – редко встречающиеся наследственные заболевания у детей. Характеризуется периодическими приступами паралича мышц скелета. Наследуется по принципу, при котором для проявления болезни достаточно одного мутантного аллеля, локализованного в аутосоме.
Выделяют три формы:
- гиперкалиемическая;
- гипокалиемическая;
- нормокалиемическая.
Гипокалиемической формой пароксизмальной миоплегии чаще всего болеют мужчины.
Первые приступы заболевания появляются с 10 лет.
Приступ возникает в утренние или ночные часы. При этом чувствуется слабость в конечностях, шее, часто доходящая до паралича. В отдельных случаях паралич распространяется на лицевую мускулатуру и дыхательные пути.
Приступ сопровождается жаждой гиперемией, потливостью. Длительность может составлять от часа до семи дней. Женщины обычно испытывают приступ в первый день менструаций.
Гиперкалиемическую форму можно встретить реже, ее приступы начинаются в младшем возрасте. Приступ провоцируют холод и длительное бездействие мышц. Он начинается с расстройства чувствительности лицевых мышц, рук и ног.
Третья форма болезни проявляется до 10-летнего возраста. Приступы проходят в течение нескольких дней или недель. Они могут быть спровоцированы низкой температурой окружающей среды, употреблением алкоголя, большими физическими нагрузками.